
Teacher
"Be ruthless with systems and be kind to people."
Michael Brooks, 1983-2020



 If you've missed the story about how BA.3.2 (a novel, divergent saltation variant) is hugely overrepresented in sequences from children, this was my original (very quick) analysis, which subsequent data extended & confirmed. 2/
If you've missed the story about how BA.3.2 (a novel, divergent saltation variant) is hugely overrepresented in sequences from children, this was my original (very quick) analysis, which subsequent data extended & confirmed. 2/

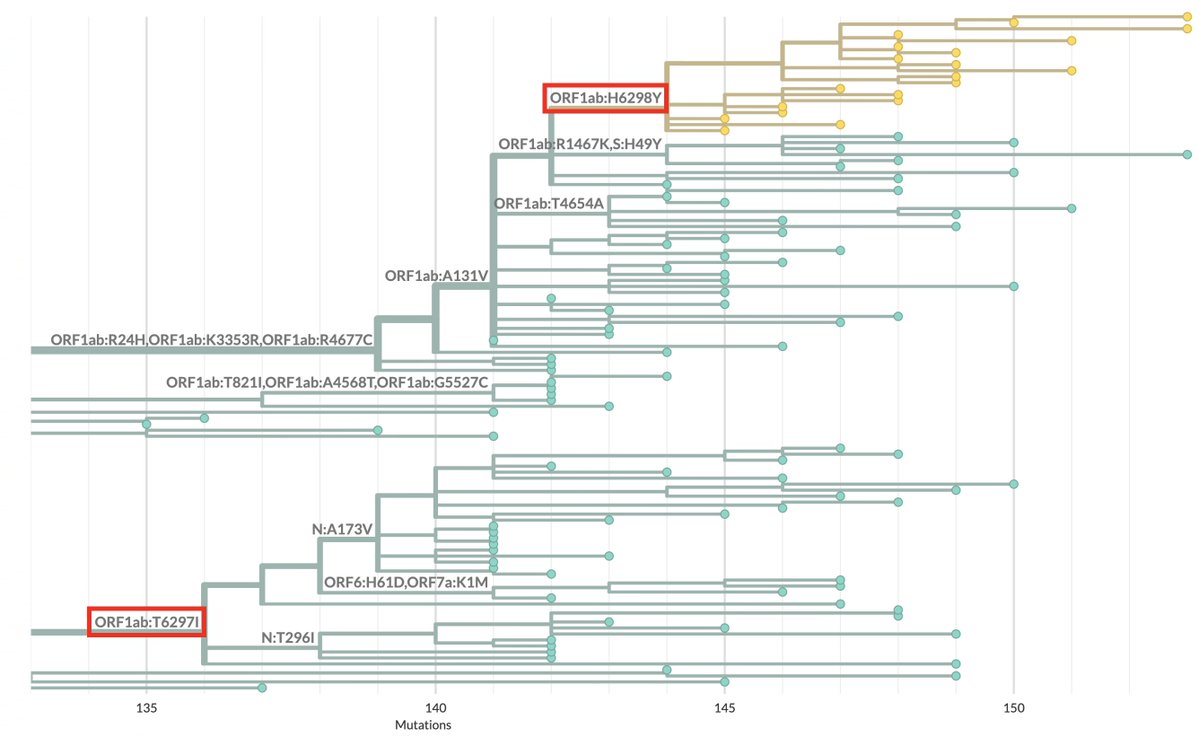
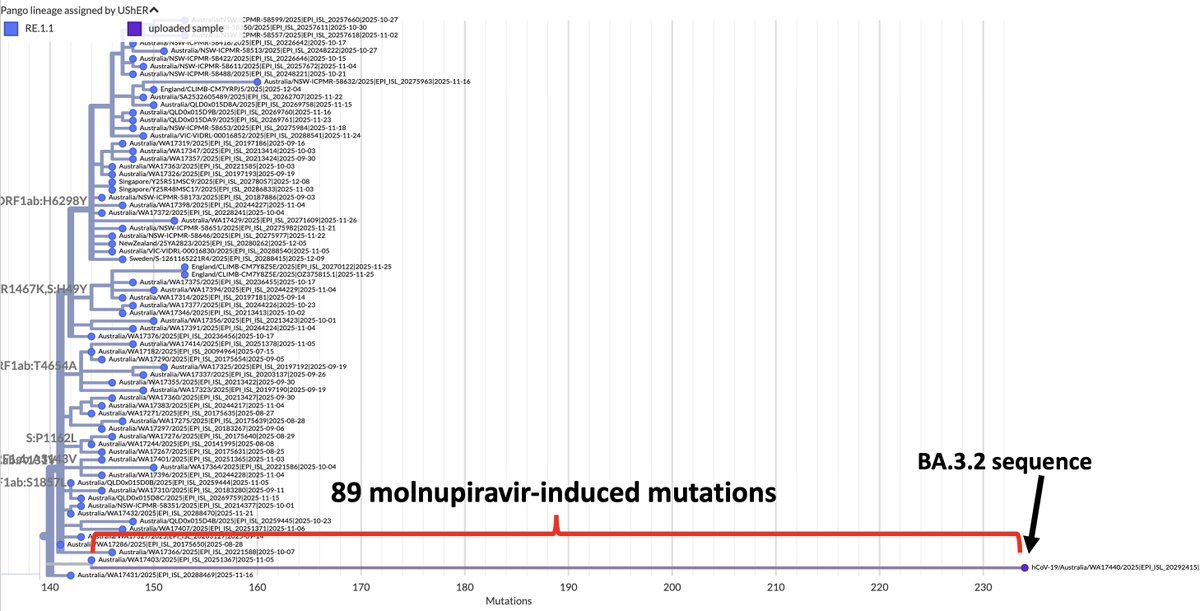 Background on MOV: It's a mutagenic drug. Its purpose is to cause so many mutations that the virus becomes unviable & is cleared. But we've long known this often does not happen. Instead, the virus persists in highly mutated form & can be transmitted. 2/
Background on MOV: It's a mutagenic drug. Its purpose is to cause so many mutations that the virus becomes unviable & is cleared. But we've long known this often does not happen. Instead, the virus persists in highly mutated form & can be transmitted. 2/
 Work by @TheMenacheryLab looked at a similar, more extensive, deletion. They deleted both QT repeats plus the next AA (∆QTQTN). In Vero cells (monkey kidney cells), it produced extra-large plaques & outcompeted WT virus—similar to furin cleavage site (FCS)-deletion mutants. 2/12
Work by @TheMenacheryLab looked at a similar, more extensive, deletion. They deleted both QT repeats plus the next AA (∆QTQTN). In Vero cells (monkey kidney cells), it produced extra-large plaques & outcompeted WT virus—similar to furin cleavage site (FCS)-deletion mutants. 2/12 
 For those not following closely, here's a 🧵 I made about BA.3.2 (not yet designated at the time) that I made some months ago, when it first burst upon the scene. 2/7
For those not following closely, here's a 🧵 I made about BA.3.2 (not yet designated at the time) that I made some months ago, when it first burst upon the scene. 2/7 In South America, this may have already happened. Recent sequences are scarce, but they nearly all have some sort of FCS-weakening mutation, mostly S:S680P in XFG.3.4.1, but with several others (S680F, S680Y, R683Q, R683W) contributing as well. 2/4
In South America, this may have already happened. Recent sequences are scarce, but they nearly all have some sort of FCS-weakening mutation, mostly S:S680P in XFG.3.4.1, but with several others (S680F, S680Y, R683Q, R683W) contributing as well. 2/4 

 First, a brief summary of the relevant parts of the preprint. They examined 30 people (from NIH RECOVER cohort) for 6 months after they had Covid, taking detailed blood immunological markers at 3 time points. 20 had Long Covid (PASC), 10 did not (CONV). 2/ biorxiv.org/content/10.110…
First, a brief summary of the relevant parts of the preprint. They examined 30 people (from NIH RECOVER cohort) for 6 months after they had Covid, taking detailed blood immunological markers at 3 time points. 20 had Long Covid (PASC), 10 did not (CONV). 2/ biorxiv.org/content/10.110…
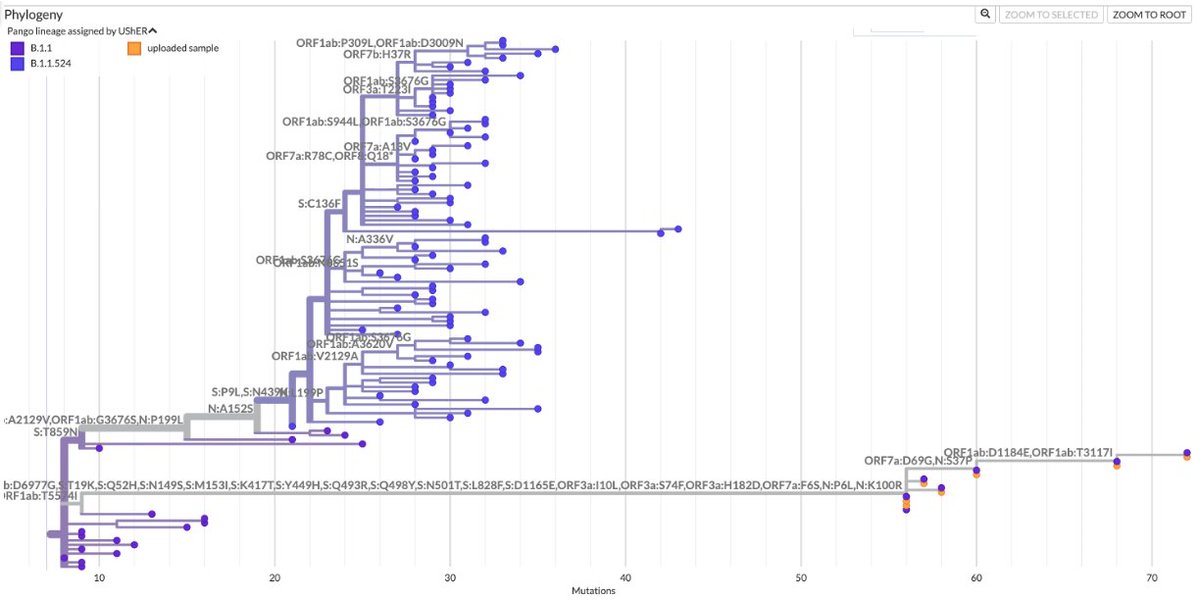
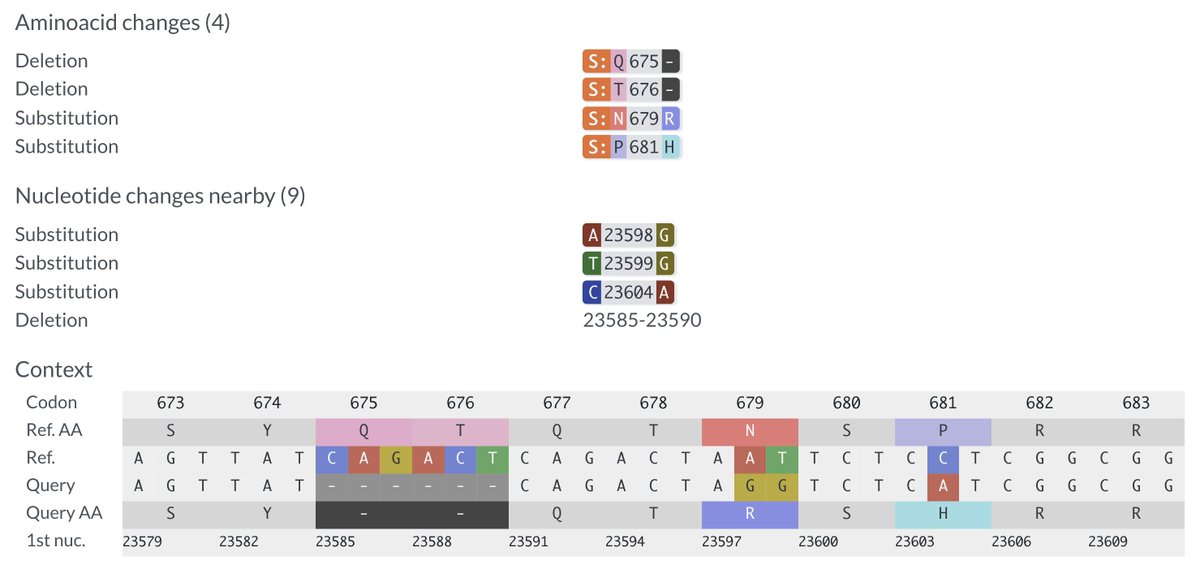
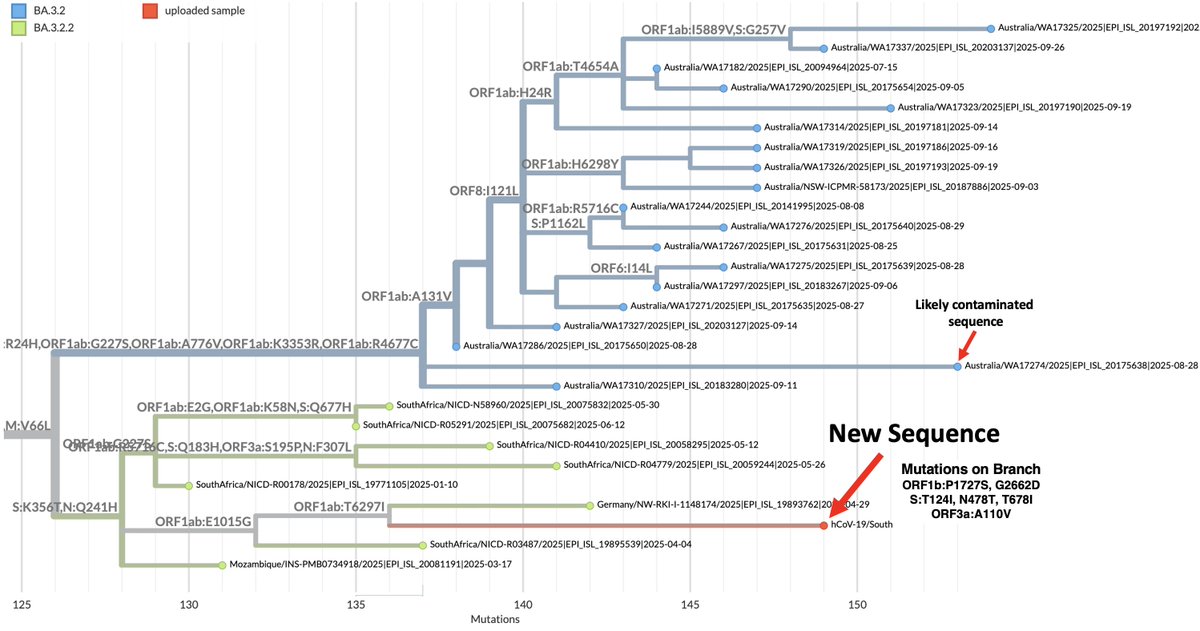
 It was collected July 15, & is most closely related to the recent S African seqs from May & June.
It was collected July 15, & is most closely related to the recent S African seqs from May & June.
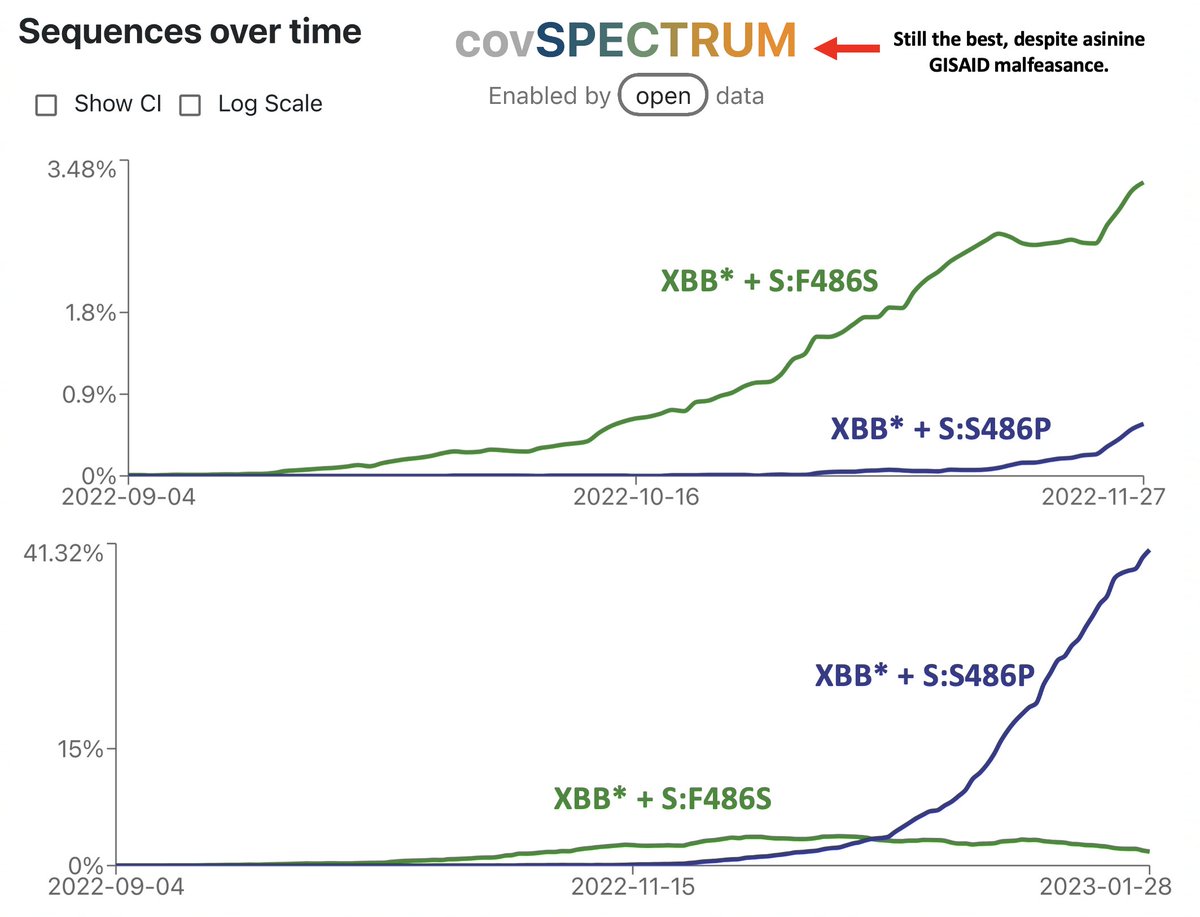
 Bottom line, in my view: BA.3.2 has spread internationally & is likely growing, but very slowly. If nothing changes, its advantage vs circulating lineages, which seem stuck in an evolutionary rut, will likely gradually grow as immunity to dominant variants solidifies... 2/9
Bottom line, in my view: BA.3.2 has spread internationally & is likely growing, but very slowly. If nothing changes, its advantage vs circulating lineages, which seem stuck in an evolutionary rut, will likely gradually grow as immunity to dominant variants solidifies... 2/9
