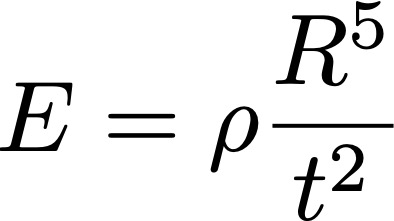Single-cell genomics assays are rooted in a handful of of technologies. They require physical isolation of cellular material, molecular barcoding, and library generation. Molecular modes include RNA, DNA (ATAC), protein and more which makes preprocessing challenging. 1/🧵 

One challenge of data (pre)processing (distinct from processing) is that multiple data types that must be processed in a manner that minimizes batch effects. The challenge is ensuring that reads generated from assays are consistently catalogued, error-corrected, and counted. 2/🧵
To address this challenge, we present cellatlas, a tool for uniform preprocessing that build on kallisto bustools (kb-python, ) and seqspec ()- a collaboration with @DelaneyKSull and @lpachter.
📖: 3/🧵github.com/pachterlab/kb_…
github.com/IGVF/seqspec/
biorxiv.org/content/10.110…
📖: 3/🧵github.com/pachterlab/kb_…
github.com/IGVF/seqspec/
biorxiv.org/content/10.110…
cellatlas is a command line tool that generates the appropriate commands to uniformly preprocess sc(n)RNAseq data, sc(n)ATACseq data, Multiome data, feature barcoding data, CRISPR (PerturbSeq) data, Spatial (Visium) data, and more. 4/🧵
cellatlas leverages the seqspec specification () to appropriately identify, extract, and correctly handle sequenced elements like barcodes and UMIs. It then selects the correct workflow (standard kallisto bustools, kITE, snATAK) to deploy. 5/🧵 biorxiv.org/content/10.110…

cellatlas is simple to use- supply:
1. sequencing reads
2. a correct seqspec specification
3. genome fasta
4. genome annotation
5. (optional) feature barcodes
and the correct workflow will be generated for you. No more worrying about providing FASTQs in the right order. 6/🧵
1. sequencing reads
2. a correct seqspec specification
3. genome fasta
4. genome annotation
5. (optional) feature barcodes
and the correct workflow will be generated for you. No more worrying about providing FASTQs in the right order. 6/🧵

cellatlas enables within-assay comparisons. We compare modes of DOGMAseq data (RNA/ATAC/Surface Protein/sample tags) from the same cell generated by R. Duerr/W. Chen. cellatlas allows us to hypothesize about an experimental cause for efficiency tradeoffs in reads/UMIs. 7/🧵

cellatlas also enables b/w assay comparisons- the challenge of which is rooted in lack of uniform preprocessing. Ascribing differences in data quality to wetlab techniques is challenging when preprocessing tools inject unnecessary variability (due to e.g. diff algorithms). 8/🧵
cellatlas solves this challenge with uniform preprocessing. Using cellatlas with 10x Multiome data (PBMCs) and DOGMAseq Multiome data (PBMCs) we find that DOGMAseq appears to be more efficient than 10x Multiome (at the same sequencing depth!) 9/🧵

We anticipate that uniform preprocessing will be useful in the development of new single-cell genomics assays, for example by revealing cross-technology tradeoffs. We also believe that uniform preprocessing will improve reproducibility. 10/🧵
This work builds on the efforts of many people including @pmelsted, @yarbslocin, @DelaneyKSull, @LambdaMoses, @lioscro, Fan Gao, @hjpimentel, @kreldjarn, @JaseGehring, Lauren Liu, @XiChenUoM, and many others. 11/🧵
cellatlas is open source and freely available: . Analysis methods can be found here: . Feedback is welcomed. 12/12github.com/cellatlas/cell…
github.com/pachterlab/BSP…
github.com/pachterlab/BSP…
@pmelsted @DelaneyKSull @LambdaMoses @lioscro @hjpimentel @kreldjarn @JaseGehring @XiChenUoM The tag should say @yarbsalocin
• • •
Missing some Tweet in this thread? You can try to
force a refresh