
I think this preprint, by @Stuartturville, is potentially one of the more important recent SARS-CoV-2 papers. It promises to unwrap some of the most mystifying aspects of Omicron surrounding cell entry, TMPRSS2 use, & reduced lung invasion/pneumonia. 1/48
https://twitter.com/StuartTurville/status/1705385838207750579
When BA.1 emerged, it was apparent it did not cause the same degree of pneumonia or disease severity as Delta (though unprecedented levels of infection resulted in similarly large numbers of hospitalizations & deaths in many areas). h/t @mike_famulare 2/48
https://twitter.com/paredesmig/status/1496594484959666176
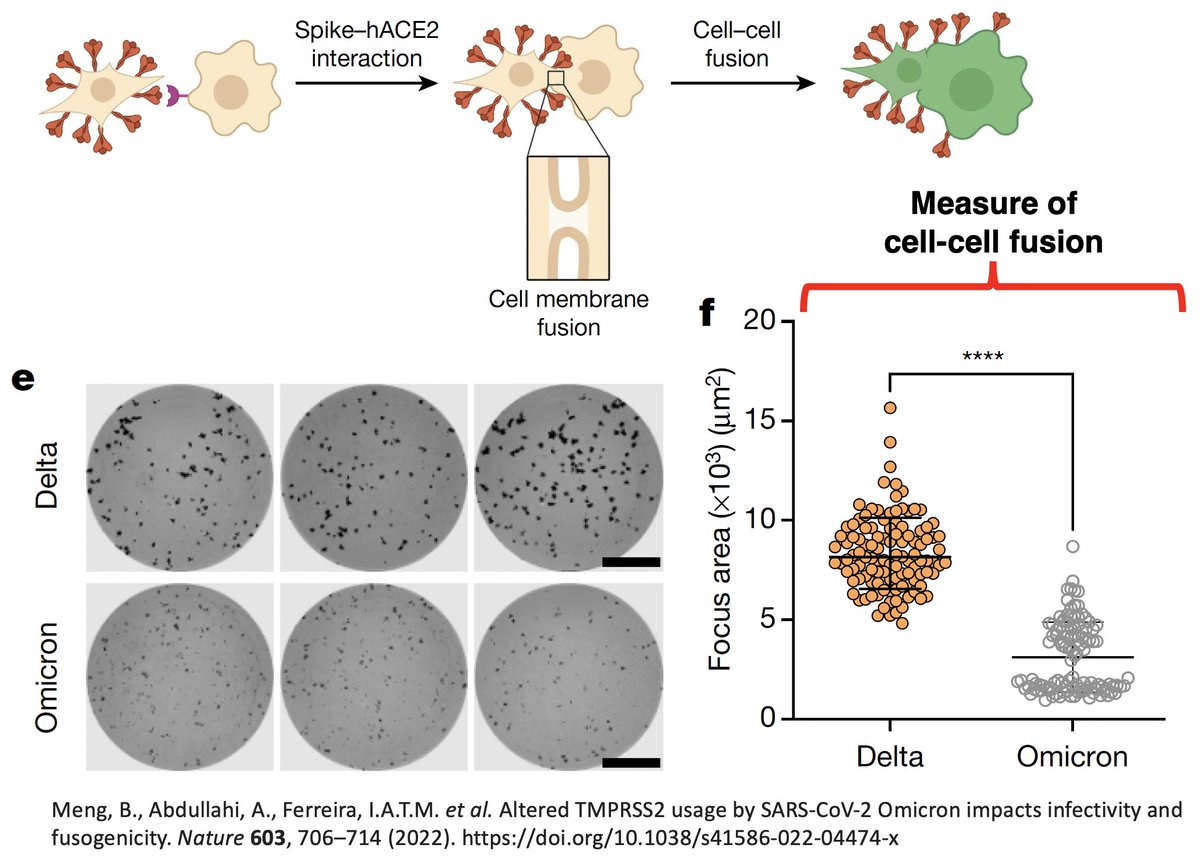
Many studies showed this was attributable to Omicron’s impaired ability to infect lung cells & reduced cell-cell fusion (syncytia formation). But what accounted for these attributes of Omicron?
(Image 1 from paper by @GuptaR_lab, @veeslerlab @sigallab @SystemsVirology) 3/48

(Image 1 from paper by @GuptaR_lab, @veeslerlab @sigallab @SystemsVirology) 3/48

A little background first: The SARS-CoV-2 spike must undergo two cleavages to efficiently infect a cell. The first cleavage comes at the S1/S2 boundary around amino acid 685 of the 1273-residue spike protein. 4/48

S1/S2 cleavage is performed by the enzyme furin in the Golgi apparatus of an infected cell. S1 & S2 remain connected but are held together by non-covalent bonds rather than much stronger covalent bonds. This optimally primes spike for later S2’ cleavage & cell entry. 5/48

The 2nd spike cleavage essential for SARS-CoV-2 cell entry occurs around 816, called S2’. There are two major ways S2’ cleavage could happen. First, 2’ can be cleaved by TMPRSS2, leading to cell membrane fusion entry. 6/48
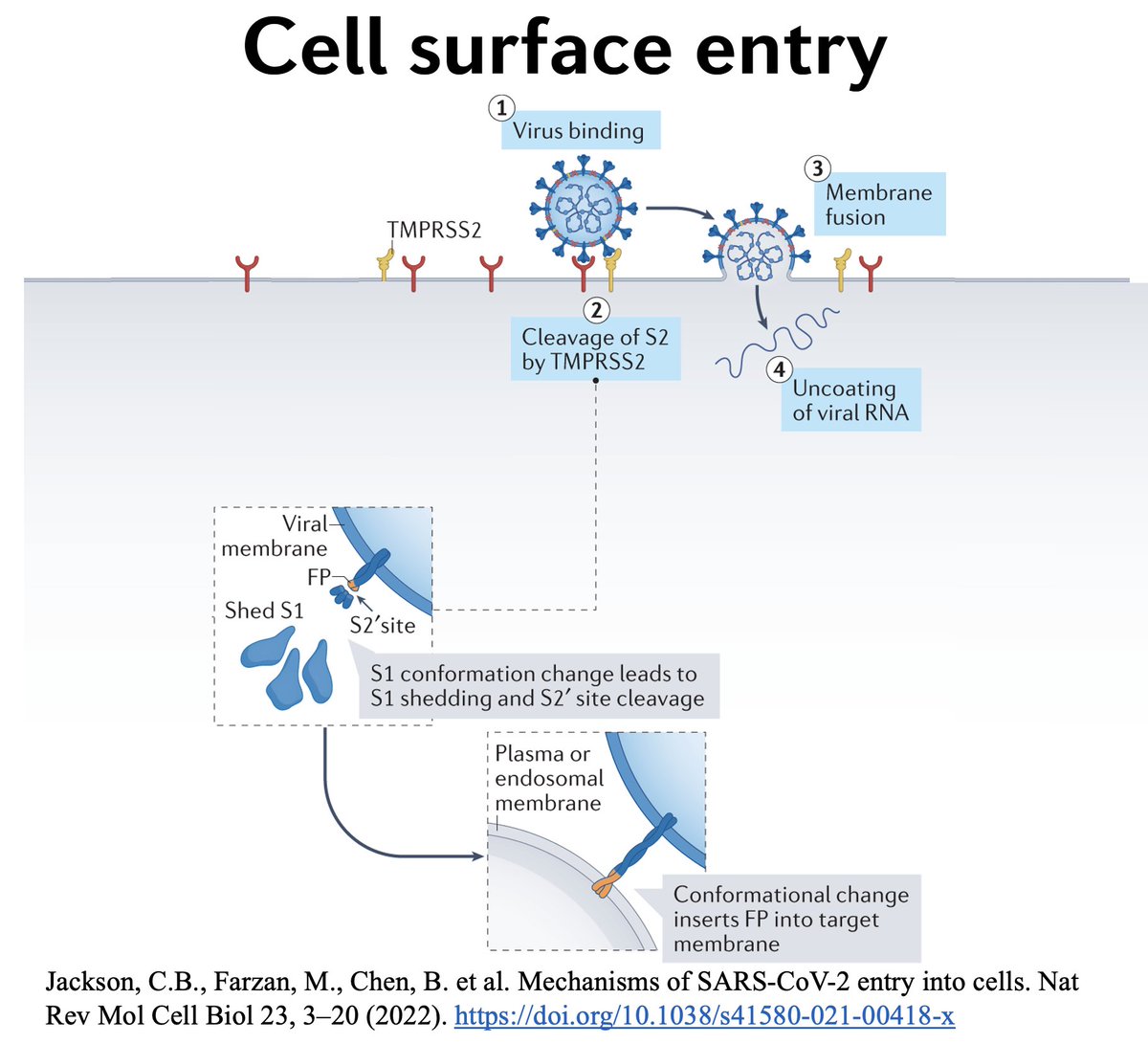
Importantly, TMPRSS2—a protein-cutting enzyme embedded in cell membranes—is especially common in lung cells, so the ability of a variant to undergo efficient S2’ cleavage by TMPRSS2 is essential for lower-respiratory tract (LRT) tropism. 7/48
Alternatively, the virus could undergo endosomal entry, in which it enters the cell enveloped in an endosome. In this case, enzymes called cathepsins cleave S2’, allowing the virus to fuse with the endosomal membrane and enter the cytoplasm, where it replicates. 8/48

After BA.1 emerged, most studies seemed to agree that its impaired ability to infect the lower respiratory tract (LRT) & fuse cells was connected to its inability to utilize TMPRSS2 for efficient spike cleavage and subsequent cell membrane fusion. 9/48




Instead, in an abrupt change from previous lineages, all of which primarily used TMPRSS2/cell membrane fusion for entry, it seemed that Omicron favored the endosomal pathway and subsequent S2’ cleavage within the endosome by cathepsins. 10/48

The endosomal-entry story neatly explained the two most striking changes in Omicron compared to previous lineages: its preference for the upper-respiratory tract (URT) & its markedly reduced cell-cell fusion capabilities.
There’s only one problem: it’s wrong.
11/48
There’s only one problem: it’s wrong.
11/48
Many studies have now shown that, in human airway cells & other models, Omicron not only utilizes TMPRSS2 but does so hyper-efficiently. See paper by @EnyaQing & Tom Gallagher that ingeniously uses viral-like particles (VLPs) & extracellular vesicles (EVs) to show… 12/48
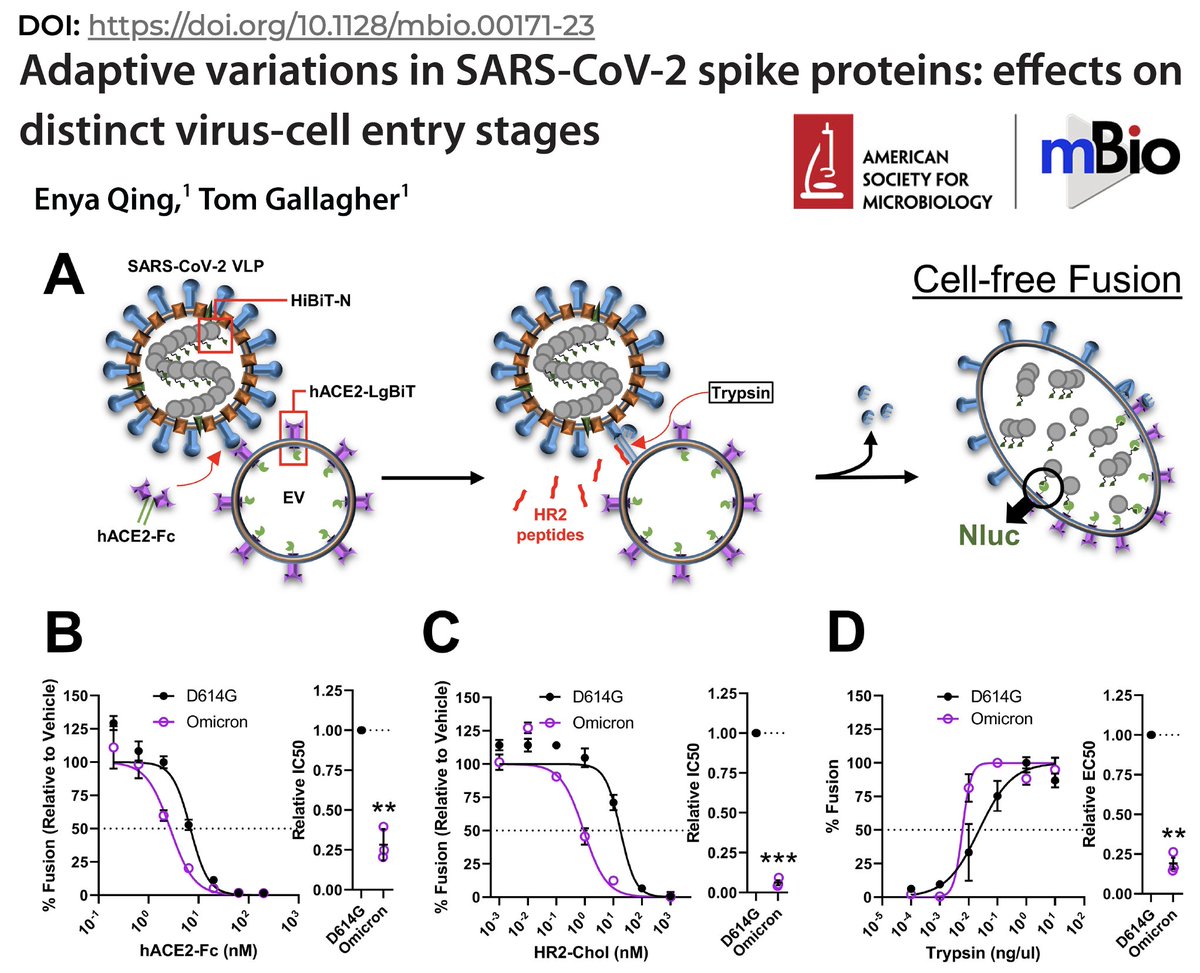
…that compared to pre-Omicron lineages, Omicron is hypersensitive to S2’ cleavage by TMPRSS2, which activates spike for fusion & cell entry. (This paper is a trove of surprising & intriguing findings, particularly the bits on S2. Deserves its own thread, really.) 13/48

Also see the earlier observation by @PeacockFlu & @wendybarclay11 that Omicron very efficiently used TMPRSS2 in human nasal cells, even as TMPRSS2 seemed almost entirely incapable of Omicron S2’ cleavage in traditional lab cell lines—now a familiar pattern. 14/48

Most recently, separate studies by @bart_haagmans & @GartnerM94 both found that in human airway organoid cells, all SARS-CoV-2 lineages, including Omicron, required TMPRSS2—but not cathepsins (used in endosomal entry) for cell entry. 15/48
https://twitter.com/GartnerM94/status/1707168610760921590
As @bart_haagmans put it in his recent study in @JVirology,
“The data indicate that the preferred SARS-CoV-2 entry route depends on the type of cell line being used.” 16/48
“The data indicate that the preferred SARS-CoV-2 entry route depends on the type of cell line being used.” 16/48

It’s now clear that in human cells, Omicron utilizes TMPRSS2 very efficiently & indeed apparently *requires* TMPRSS2 for successful infection. Endosomal entry seems an artifact confined to lab cell lines, many of which overexpress TMPRSS2 & ACE2. 17/48

But if Omicron S2’ cleavage by TMPRSS2 is ultra-efficient, leading to successful cell membrane-fusion entry—strongly implying that endosomal entry plays no major role—why have Omicron lineages been so unsuccessful at infecting lung cells, which have TMPRSS2 in spades? 18/48
It was all an utter mystery to me.
Until I read the new paper by @StuartTurville & @milogiannakis. In it, they provide a compelling explanation for the contradictory findings regarding Omicron & TMPRSS2 & for Omicron’s reduced LRT tropism. 19/48
Until I read the new paper by @StuartTurville & @milogiannakis. In it, they provide a compelling explanation for the contradictory findings regarding Omicron & TMPRSS2 & for Omicron’s reduced LRT tropism. 19/48
https://twitter.com/StuartTurville/status/1705392126912266616
When BA.1 emerged, labs struggled to grow it in cell lines they’d successfully used to grow pre-Omicron variants: “In many cases… the ACCE2-TMPRSS2 pathway…appeared no longer functional for Omicron lineages.” So Turville engineered a cell line—VeroE6-T2 (E6-T2)— 20/48

—in which all variants, including Omicrons, replicated well. Puzzlingly, E6-T2 highly expressed TMPRSS2, which in previous studies (& all other cell lines Turville tested) potently inhibited Omicron replication. Somehow, in contrast to other engineered cell lines… 21/48
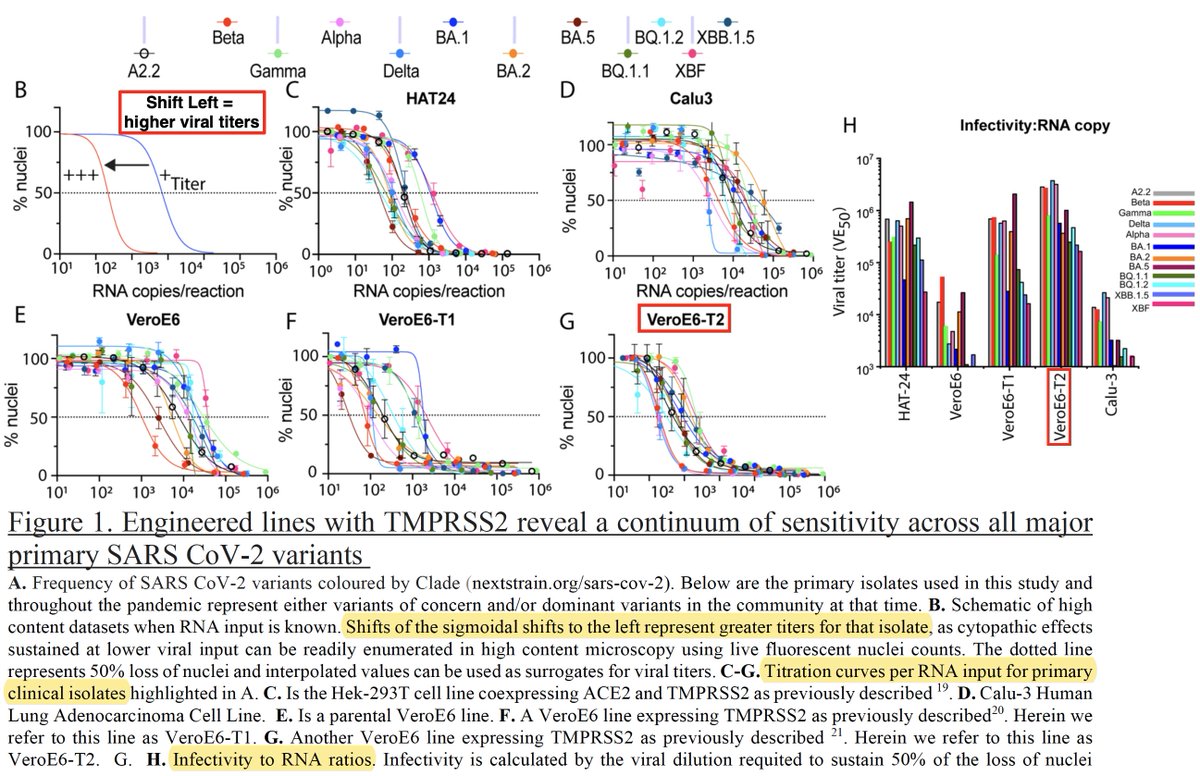
…rich in TMPRSS2, Omicron S2’ cleavage successfully takes place in E6-T2 cells. Turville proved Omicron utilized TMPRSS2 by showing that a TMPRSS2 inhibitor (Nafamostat) stifled Omicron replication while a compound that prevents endosomal cell entry (E64D) had no effect. 22/48

Exactly why Omicron succeeds in E6-T2 cells but not other high-TMPRSS2 cell lines remains a mystery (as far as I can tell). Their next experiment, however, was even more confounding. VeroE6-T2 cells have low levels of ACE2… 23/48

Since ACE2 is the essential cell-membrane receptor used by all SARS-CoV-2, higher ACE2 expression should increase viral cell entry & replication, & this is what previous studies have found. So Turville created an E6-T2 cell line with high levels of ACE2, (E6-T2-ACE2). 24/48

As expected, increased ACE2 levels enhanced replication—in pre-Omicron variants. But for Omicron, it potently *inhibited* replication. Even within Omicron & pre-Omicron lineages, there’s a distinct downward trend in viral titers in high-ACE2+TMPRSS2 cells—note log scale. 25/48

What’s going on here? Why in the world would increasing the density of a virus’s receptor reduce its ability to infect a cell? Quite the riddle. 26/48
To understand the (partial) answer, we need to know that TMPRSS2 can cleave ACE2. (I felt rather stupid for not knowing this.) It sounds as if this might destroy its usefulness as a receptor, but ACE2 actually *requires* this cleavage to perform its function in the… 27/48
…renin-angiotensin-aldosterone system (RAAS), which regulates blood pressure & electrolyte balance. TMPRSS2 cleavage preserves ACE2 in the cell membrane by competing with a different enzyme (ADAM17) that cleaves ACE2 in a way that chucks it off the membrane altogether. 28/48

Two great papers, one by Tom Gallagher & Stanley Perlman & one by @snpoehlm, showed TMPRSS2-mediated ACE2 cleavage potently enhanced SARS-CoV-1 replication—30-fold increase in cell entry, 9-fold increase in viral RNA—much like in pre-Omi SARS-2 variants. 29/48


But ACE2 also plays an entirely different role in the body: pairing up with one of two solute-carrier proteins—B0AT1 (also called SLC6A19) or SIT1 (SLC6A20) to chaperone certain amino acids across the plasma membrane & into cells. 30/48

In fact, that vast majority of ACE2 in the body acts in this chaperone role rather than its more well-known RAAS role. How is this relevant? ACE2 in its chaperone role, attaches to B0AT1 or SIT1 in a way that conceals its cleavage site, preventing TMPRSS2 from cutting it. 31/48

In a 2020 paper, the man who discovered & first described the function of B0AT1, Bruce R. Stevens, wrote a fantastic preprint describing how the B0AT1-ACE2 chaperone complex hides the ACE2 cleavage site from TMPRSS2 in all conformations. 32/48

Now we finally seem to have the beginnings of an explanation for why Omicron’s strikingly reduced ability to invade the lungs: the high LRT levels of TMPRSS2 efficiently cleave ACE2, which improves pre-Omi cell entry & replication but potently inhibits Omicron variants. 33/48
Turville provided further support for this hypothesis by putting an uncleavable version of ACE2 in the high-ACE2+TMPRSS2 E6-T2-ACE2 cells that Omicron previously failed in. Result: Omicron replication completely rescued & now equal to pre-Omicron variants. 34/48

Since intestinal ACE2 overwhelmingly functions in its amino acid-transport chaperone role (which cannot be cleaved by TMPRSS2), one would expect intestinal replication of Omicron to be as efficient as pre-Omicron variants, in contrast to the attenuation seen in lungs. 35/48
Wastewater SARS-CoV-2 levels, which presumably reflect intestinal replication, seem to confirm this. WW levels at the peak the Omicron and Delta waves correspond well to estimated case prevalence based on both seroprevalence & official case numbers. 36/48

This paper casts a clarifying light on the crucial & confounding mystery of why Omicron fails to succeed in lung cells despite its demonstrated efficiency at utilizing TMPRS2. It appears a real breakthrough to me that deserves more attention than it seems to have received. 37/48
Many mysteries remain. Why exactly does ACE2 cleavage by TMPRSS2 enhance pre-Omicron lineage infection? And how does this cleavage inhibit Omicron? The most obvious explanation is that Omicron’s extremely divergent spike cannot bind to cleaved ACE2. But this can’t be right. 38/48
Omicron successfully infected Vero-E6-T2 cells, which had high levels of TMPRSS2 & low levels of ACE2 was drastically inhibited in E6-T2 cells with high levels of both TMPRSS2 & ACE2. If Omicron’s failure was due to an inability to utilize cleaved ACE2… 39/48
… then it ought to have done better in the high-ACE2+TMPRSS2 cells, where a small amount of uncleaved ACE2 might have slipped through. But the opposite happened. There seems to be something about the absolute amount of ACE2 cleavage itself that stymies Omicron. 40/48
High ACE2 affinity seems to result in greater inhibition in TMPRSS2 + cleavable ACE2 cells compared to the same cells w/non-cleavable ACE2. Is this a clue that could lead us to an explanation?
A million things are going on in cells, so the explanation is likely complex. 41/48
A million things are going on in cells, so the explanation is likely complex. 41/48
Another major mystery to me is why Omicron is able to replicate so well in human nasal epithelial cells (hNECs). ACE2 acts in its RAAS role there, as in the lungs, so it ought to be cleaved by TMPRSS2. Yet Omicron seems proficient at using TMPRSS2 in hNECs. 42/48

@GartnerM94’s study is suggestive on this question. First, note that Omicron is actually outcompeted by WT & Delta in nasal cells (& other respiratory cell tested) 2 days post-infection (dpi). But Omicron somehow gets a jump start on the others—as in @PeacockFlu’s study. 43/48


Furthermore, while the TMPRSS2 inhibitor Camostat annihilated infection by WT & Delta, Omicron replication was only mildly reduced. But a compound inhibiting endosomal entry (CA-074 Me) had ~zero effect on all variants, so this can’t explain the discrepancy. 44/48

Gartner suggests Omicron may be utilizing a different protease for S2’ cleavage in hNECs. If so, in addition to explaining Omicron’s success in the face of TMPRSS2 inhibition, might this somehow also explain its remarkably rapid replication in the first hours of infection? 45/48

Omicron doesn’t fuse cells (form syncytia) as much as previous variants. One merit of the endosomal-entry hypothesis was that it readily explained this. In endosomal entry, membrane fusion & occurs in endosomes within the cell, not on the cell membrane. 46/48

But now that endosomal entry seems disproven and especially given findings that Omicron may actually be much *more* primed for fusion than previous variants, its reduced syncytia formation again begs for an explanation. (@EnyaQing & Tom Gallagher again below.) 47/48

I’m sure others have far more insight on these questions than I do.
In any case, it’s exciting to see some light being shed on the various Omicron enigmas, & I hope others build on Turville’s insights & illuminate more of Omicron’s dark secrets. 48/48
In any case, it’s exciting to see some light being shed on the various Omicron enigmas, & I hope others build on Turville’s insights & illuminate more of Omicron’s dark secrets. 48/48
• • •
Missing some Tweet in this thread? You can try to
force a refresh







