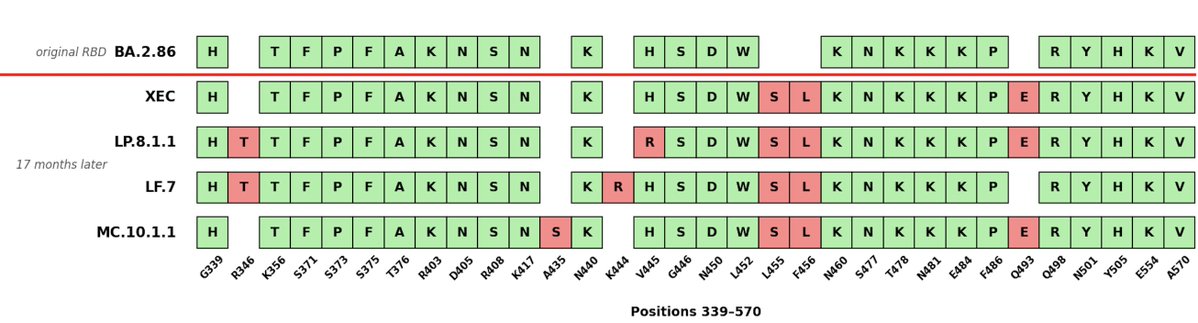
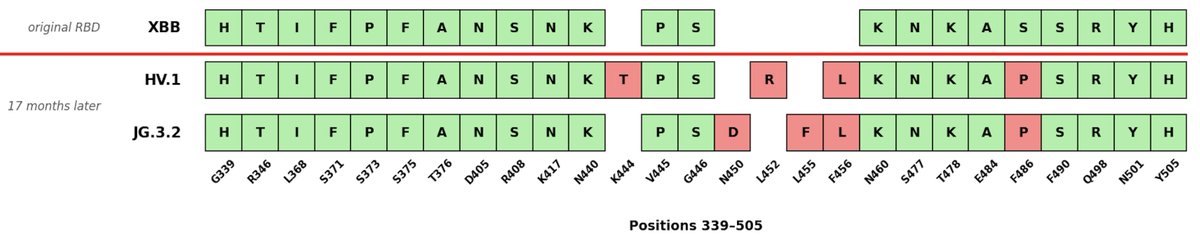
We are now officially into year 5 of the SARS-CoV-2 pandemic/endemic.
I gave a lecture to my virology class this Fall about the history of the pandemic through the lens of viral genotypes.
I thought I would share that lecture as a thread.
This is a long one.
1/
I gave a lecture to my virology class this Fall about the history of the pandemic through the lens of viral genotypes.
I thought I would share that lecture as a thread.
This is a long one.
1/
In the beginning there were 2 genotypes of SARS-CoV-2, A and B.
The two differed by only 2 nt, but both lineages would go on to circle the globe.
2/
The two differed by only 2 nt, but both lineages would go on to circle the globe.
2/

The fact that both lineages were present in the Wuhan Seafood market from very early on is one strong piece of evidence that the market was the likely origin.
If the market were just a single superspreader location, you wouldn’t have expected it to have both lineages.
3/
If the market were just a single superspreader location, you wouldn’t have expected it to have both lineages.
3/
The virus was found to be closely related (96% identical) to a bat sarbecovirus, RaTG13.
A striking difference was a 4 AA insertion that would create what is called a furin-cleavage site (FCS), a protein sequence that could be cut by the cellular protein furin.
4/
A striking difference was a 4 AA insertion that would create what is called a furin-cleavage site (FCS), a protein sequence that could be cut by the cellular protein furin.
4/

Lab leak proponents will claim that this is evidence that the virus was engineered because many other coronaviruses have an FCS at the same site, and investigators had talked about testing these kind of changes.
5/
5/
Zoonosis proponents will counter that coronaviruses make random insertions all the time, and no idiot would generate an FCS that was preceded by a proline (P) since that would make a very poor cleavage site.
6/
6/
The virus agreed with the zoonosis proponents on this account and proceeded to eliminate the Proline numerous times. 681P went extinct in circulating lineages years ago.
7/
7/
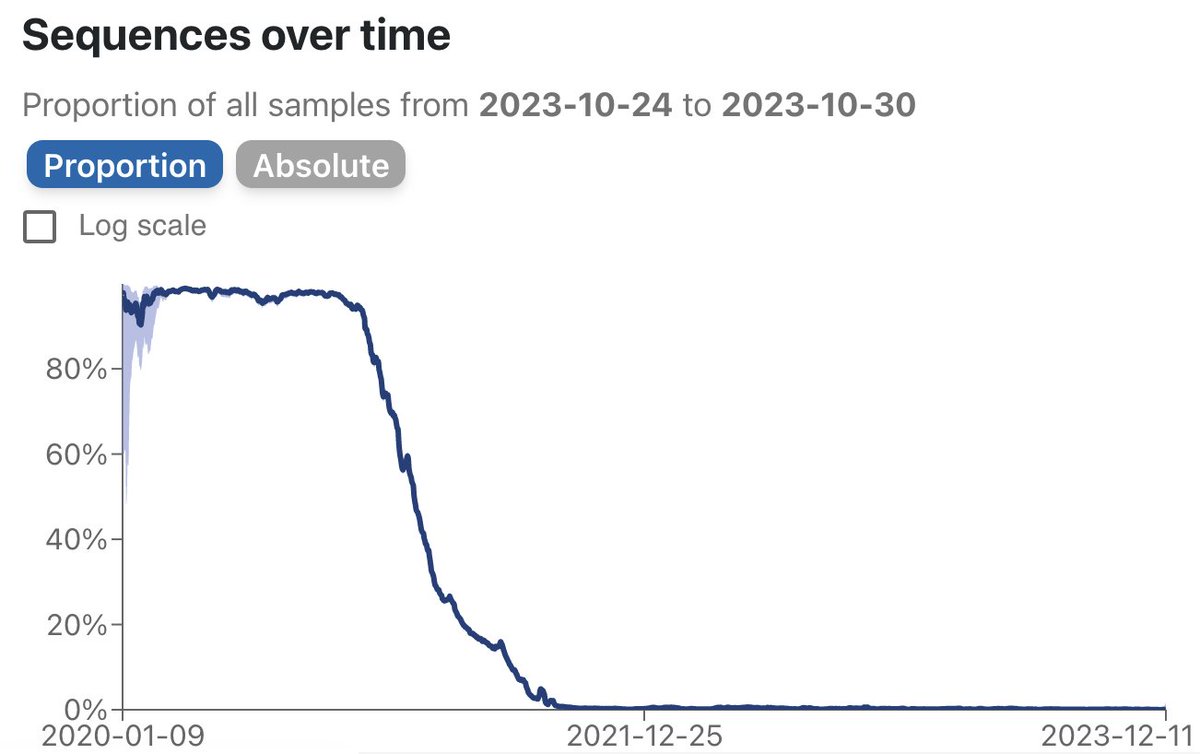
But the A and B lineages started having offspring and eventually a B descendant called B.1 took over. Bette Korber was the first to point out the dramatic increase in lineages containing the mutation D614G, a key mutation in the B.1 lineage.
8/
8/

I now need to explain how PANGO designations work if you aren’t familiar already.
Every lineage has a numerical designation starting with A or B.
Any time they get a descendant, they get the same designation as the parent with a new number added at the end.
9/
Every lineage has a numerical designation starting with A or B.
Any time they get a descendant, they get the same designation as the parent with a new number added at the end.
9/
The first descendant of B was B.1, the second was B.2, etc.
When B.1 had descendants, they were B.1.1, B.1.2, etc
And so on.
10/
When B.1 had descendants, they were B.1.1, B.1.2, etc
And so on.
10/
However, they couldn’t have strings of numbers going on forever, so they put a cap at 3 numbers.
Once a lineage gets a 4th number, that is converted to the next available letter in the alphabet.
11/
Once a lineage gets a 4th number, that is converted to the next available letter in the alphabet.
11/

The first time this happened was with the lineage B.1.1.1.1, which became C.1.
Eventually they ran out of single letters and had to switch to a two-letter code. We are about halfway towards needing a 3-letter code.
12/
Eventually they ran out of single letters and had to switch to a two-letter code. We are about halfway towards needing a 3-letter code.
12/
Last thing, when a viral recombination occurs, the lineage starts with X (like XBB), but then all the same rules apply.
BTW, I would like to thank the dedicated scientists (mostly volunteers) that keep track of these lineages through tireless analysis. The list is long.
13/
BTW, I would like to thank the dedicated scientists (mostly volunteers) that keep track of these lineages through tireless analysis. The list is long.
13/
The B.1 and B.1.1 lineages dominated by mid-2020 and everyone thought the pandemic would soon be over.
Then something surprising (to me) happened with a virus that is supposed to make virus very few replication errors.
14/
Then something surprising (to me) happened with a virus that is supposed to make virus very few replication errors.
14/
We started getting various lineages that seemed to be spreading at a much faster rate. The lineages (at the time) were called the UK variant (B.1.1.7), the South Africa variant (B.1.351), and the Brazil variant (B.1.1.28.1/P.1). These are now called Alpha, Beta, and Gamma.
15/
15/
All three of these lineages, which were from very different viral backgrounds and parts of the world, had the same Spike mutation N501Y.
Why did this only start appearing a year into the pandemic?
16/
Why did this only start appearing a year into the pandemic?
16/

Some said it was about immune evasion, and the virus didn’t need it before because no one had immunity before.
I never bought this. The most successful of the three N501 lineages was Alpha, which is not particularly immune evasive.
17/
I never bought this. The most successful of the three N501 lineages was Alpha, which is not particularly immune evasive.
17/
Others said that N501Y gave a general growth advantage because it enhanced receptor binding.
If true, why did it take so long to be select for it?
18/
If true, why did it take so long to be select for it?
18/
The answer of the timing of N501Y lineages probably has to do with how they emerged.
There is a lot of evidence that all 3 of these lineages were derived from persistent infections. This helps explain the timing.
19/
There is a lot of evidence that all 3 of these lineages were derived from persistent infections. This helps explain the timing.
19/
We know now that people can be infected for months or even years with SARS-CoV-2 in some instances, and this is basically like sending the virus to college.
20/
20/
In a persistent infection the virus has lots of time to try out different combinations of changes that it doesn’t get to try when it is ‘working’ (in circulation).
I know I’m anthropomorphizing, but it fits.
21/
I know I’m anthropomorphizing, but it fits.
21/
Generally, the novel lineages from persistent infections take months to years before they ‘escape’ and start circulating again. (In the vast majority of cases, this never happens, it just stays in the one patient)
22/
22/
Although the Alpha/Beta/Gamma lineages all had N501Y, they also had other changes (such as changes at the FCS), and they were all B.1/B.1.1 derivatives (containing D614G).
23/
23/
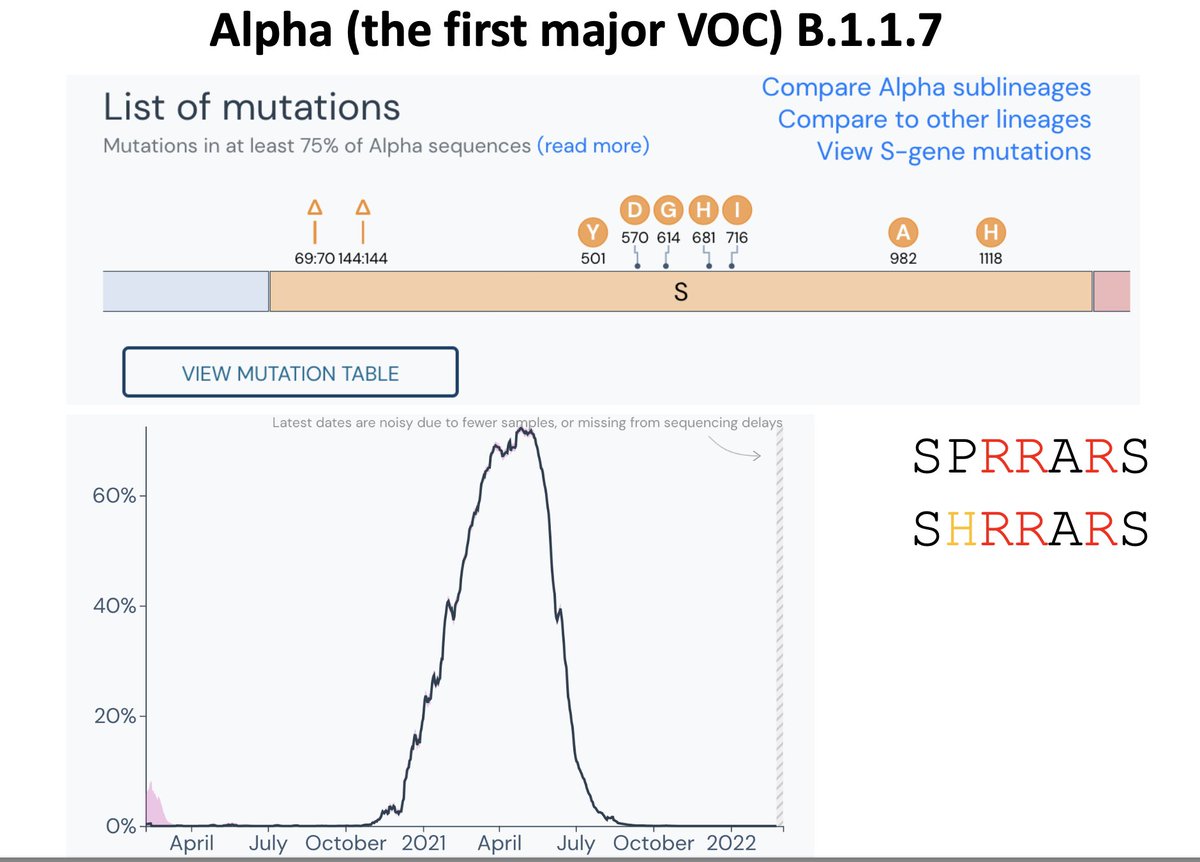
It was probably just a matter of timing.
In the first few months B.1/B.1.1 took over, started a bunch of persistent infections, and then three of these started circulating again a few months later, and they all had certain ‘obvious’ changes like N501Y.
24/
In the first few months B.1/B.1.1 took over, started a bunch of persistent infections, and then three of these started circulating again a few months later, and they all had certain ‘obvious’ changes like N501Y.
24/
Opps, I guess twitter has a string limit now. I'll continue in another thread.
• • •
Missing some Tweet in this thread? You can try to
force a refresh