Aristotle was the first to notice honeybees dancing. In 1927 Karl von Frisch decoded the waggle. How it works was "explained" by MV Srinivasan AM FRS in the 1990s. Except @NeuroLuebbert found his papers are junk. A 🧵 about her discovery & our report: 1/arxiv.org/abs/2405.12998
First, if you're not familiar with the waggle, it's Nature magic! Watch this video for cool footage and an introduction.
Aristotle's observations in Historia animalium IX are arguably one of the first instances of observation driven inquiry and science. 2/
Aristotle's observations in Historia animalium IX are arguably one of the first instances of observation driven inquiry and science. 2/
Karl von Frisch decoded the waggle, meaning he figured out how the number of waggles, and their direction, communicate information about the distance and direction of food sources. von Frisch won the Nobel Prize for his discovery. But exactly how it works remained a mystery. 3/ 

BTW von Frisch was part Jewish, and the Nazis accused him of working with too many foreigners, too many women, and practicing "Jewish science". He was first classified as 1/8 Jewish which let him keep his post, but ultimately reclassified to 1/4.. a story for another thread.. 4/
In the 1990s, MV Srinivasan started writing papers purporting to explain the mechanisms underlying the waggle. These papers made him famous. He is a member of the Royal Society. He won the Prime Minister's Prize for Science, etc. etc. 5/en.wikipedia.org/wiki/Mandyam_V…
In 2020 @NeuroLuebbert, at the time a new PhD student, rotated in a lab where she was assigned two Srinivasan papers to present. New to the topic, she read some additional papers for context. She noticed the same data.. appearing again and again.. in different experiments. 👀 6/
In a blog post, she tells the story of the reaction she received when she pointed this out to her (tenured) professor at the time, and to others. She was basically told not to waste her time: "a lot of the scientific literature has problems". 7/liorpachter.wordpress.com/2024/07/02/the…
She tweeted out her discovery at the time. It seemed to her like a pretty big deal. It was. The response she got was basically a collective shrug. Almost no likes or retweets. 8/
https://x.com/NeuroLuebbert/status/1266162740218888192
But one of the responses put her in touch with @MicrobiomDigest, and led to two @PubPeer comments. See
You can guess what happened with these comments. Bupkes. 9/pubpeer.com/publications/F…
You can guess what happened with these comments. Bupkes. 9/pubpeer.com/publications/F…
I found out about this from @NeuroLuebbert years later after she had joined my lab. To make a long story (told in the blog post ) we went back to the papers (half a dozen) and noticed many additional problems. We decided to write up her observations. 10/ liorpachter.wordpress.com/2024/07/02/the…

What we found was very, very bad. For example, six papers reported R^2 = 0.99.. These are fits to data from experiments tracking live animals on primitive cameras in the early 2000s. 🤔 11/

This is an example with reported r^2 = 0.999 for data that obviously isn't. It's from a PNAS paper
Maybe just a typo? Well no. The papers are filled with ridiculous r^2 values, and in our report we perform analyses showing they just can't all be real. 12/ pnas.org/doi/full/10.10…
Maybe just a typo? Well no. The papers are filled with ridiculous r^2 values, and in our report we perform analyses showing they just can't all be real. 12/ pnas.org/doi/full/10.10…

@NeuroLuebbert also found more duplications and manipulations. Crazy stuff. Identical data reported for different experiments across different papers. Some people think this kind of work, which @MicrobiomDigest does, is fun sport. It isn't. It's terribly depressing. 13/

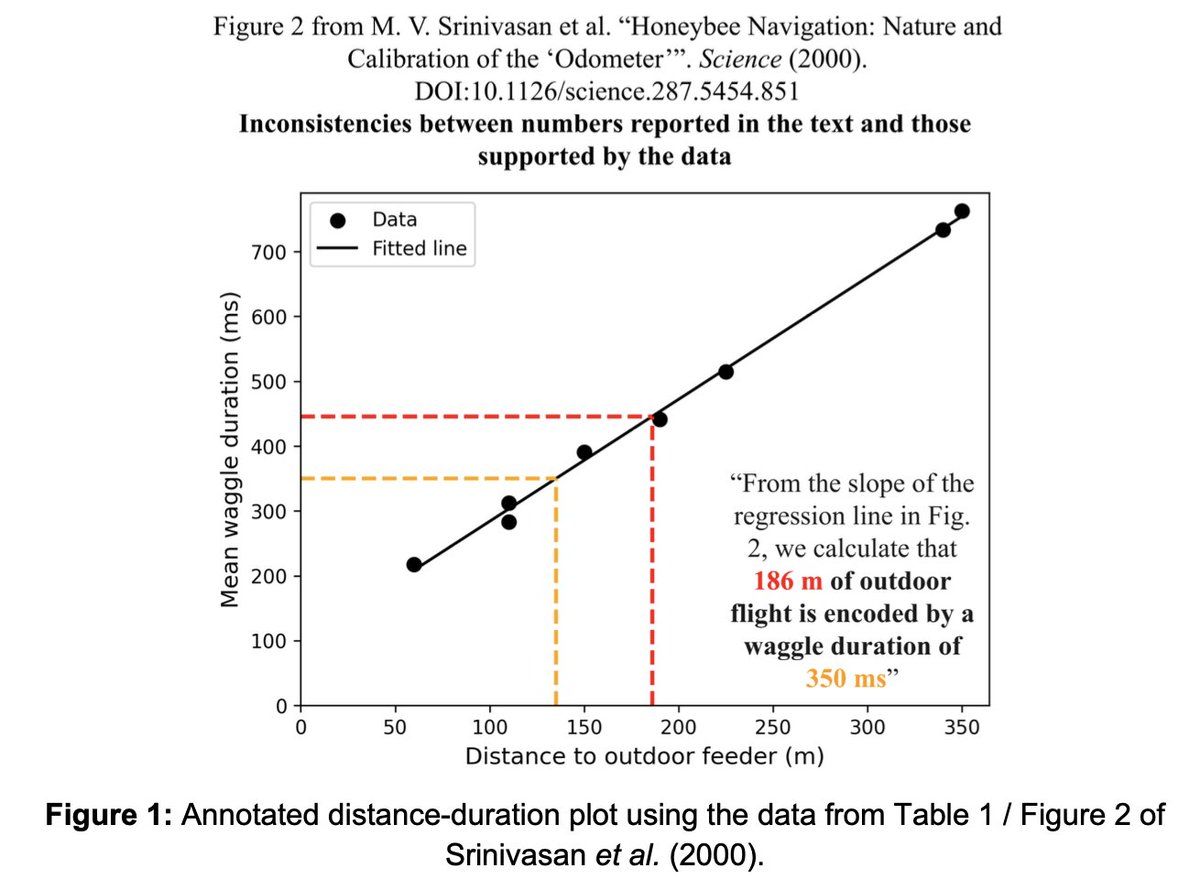
Laura was brilliant at digging through this junk. She didn't just find duplications, but also errors in key results. One is super important, a key calibration from a regression that is done incorrectly by Srinivasan et al, and inconsistent with work of others, e.g. @schuemaa. 14/

I've mentioned MV Srinivasan several times, but we didn't set out to find errors in his work. His name was just the only one present on all the papers we found problems with. We haven't gone through his whole corpus of work 😬. 15/scholar.google.com/citations?user…
We submitted our report to @biorxivpreprint who rejected it. I get it. They didn't view it as "research", and I get where the policy comes from. But it was frustrating. Especially to be told we our manuscript contained "content with ad hominem attacks". It didn't. It doesn't. 16/
So we sent it to @J_Exp_Biol, where some of the papers were from. They rejected it as well, and asked us to individually contact all the journals. This was even more frustrating. The whole here is much greater than the sum of the parts. 17/
Eventually @J_Exp_Biol did post corrections to two of Srinivasan's papers published with them that @NeuroLuebbert had flagged. In this one Srinivasan *believes* everything is ok. 18/ journals.biologists.com/jeb/article/22…
In this one he talks about his paper *likely* containing the correct values.
Are publishers now assigning likelihood of truth?Practicing belief based science? @J_Exp_Biol's corrections here are very weak sauce. 19/journals.biologists.com/jeb/article/22…
Are publishers now assigning likelihood of truth?Practicing belief based science? @J_Exp_Biol's corrections here are very weak sauce. 19/journals.biologists.com/jeb/article/22…
Eventually we submitted our report to @arxiv. Although even they flagged and held it for 2 weeks, we're grateful to them for posting it. But Science has a big problem. There ought to be a place to publish critique of a body of work. Not just a complaint about one paper. 20/
That's why we titled our blog post "The Journal of Scientific Integrity". Where is this journal? Why is it so taboo to face misconduct by a scientist? People are eager to pounce on women (e.g. Claudine Gay). But I don't expect Srinivasan to be featured in the @nytimes. 21/
And yet, this is a very serious matter. I do believe that the vast majority of errors in science are innocent mistakes. But when they appear not to be, it should be ok to speak up. It should be ok to publish a critique on a body of work. 22/
Right now it's not. But it is, apparently, ok to always present "perfect" data and regressions where all the points lie exactly on a line. Srinivasan is still presenting r^2=0.99. This talk is from just a few years ago at the #ICRA18 plenary. 23/
Truth matters everywhere, but if we lose it in science, then there is meager hope elsewhere. In our abstract we conclude that "[our investigation] suggests that redoing the experiments in question is warranted." Hopefully that will happen.
Kudos to @NeuroLuebbert. 🐝 24/24
Kudos to @NeuroLuebbert. 🐝 24/24
• • •
Missing some Tweet in this thread? You can try to
force a refresh