
SKANDAL: " Wurden die Chargen EM0477 und EJ6788 vom deutschen Markt als Todes - Chargen zurück gerufen ohne Deklaration gegenüber der Öffentlichkeit?"
Sehen wir uns die erste Charge EM0477 nach den Daten bei VEARS an:
234 Angehöriger und Ärzte meldeten nach Verabreichung von der Charge EM0477 den Tod nach dieser Impfung
148 Menschen meldeten dauerhafte Schwerbehinderung nach Verabreichung der Charge EM0477
109 Menschen deklarierten lebensbedrohliche Erkrankungen nach Verabreichung von EM0477
513 Ärzte meldeten Patienten nach Verabreichung der Charge EM0477, bei denen eine Behandlung im Krankenhaus erforderlich wurde
1.218 weitere Menschen meldeten weitere gesundheitliche Schäden nach Verabreichung on EM0477
und jetzt kommt es 79 Impfzentren und Ärzte sowie Angehörige meldeten sofort den Tod nach Verabreichung von EM0477. Das ist krass.
Sehen wir uns die nächst Charge an EJ6788
152 Angehöriger und Ärzte meldeten nach Verabreichung von der Charge EJ6788 den Tod nach dieser Impfung
38 Menschen meldeten dauerhafte Schwerbehinderung nach Verabreichung der Charge EJ6788
64 Menschen deklarierten lebensbedrohliche Erkrankungen nach Verabreichung von EJ6788
357 Ärzte meldeten Patienten nach Verabreichung der Charge EJ6788, bei denen eine Behandlung im Krankenhaus erforderlich wurde
651 weitere Menschen meldeten weitere gesundheitliche Schäden nach Verabreichung von EJ6788
und jetzt kommt es 30 Impfzentren, Ärzte und Angehörige meldeten sofort nach Verabreichung von EJ6788 den Tod dieser Menschen.
Wir glichen die beiden Chargen mit unserer Statistik ab. Die beiden vorstehenden Chargen übersteigen mindestens um das 5fach die Top 1 - Chargen in unserer eigenen Kanzlei Statistik, was Tote und Verletzte angeht. Für die erste Charge erklärten bisher aber nur 2 Mandanten, dass diese Charge schadenursächlich sei und für die 2 Charge EJ6788 nur 3 weitere Mandanten, dass diese Charge die Ursache für Impfschäden gesetzt habe. Die Statistik von VEARS und unsere Statistik divergiert gigantisch, weshalb es nur einen logischen Schluss gibt:
Die Charge EM0477 und die Charge EJ6788 wurden vom Markt durch Rückruf zurück genommen und zwar klammheimlich ohne irgendein Aufsehen zu erregen. Anders ist die Divergenz nicht erklärbar.
Anliegend in den Bildern sind die Top 5 aufgelistet. Jede dort angegebene Charge ist signifikant weniger schadensträchtig als EM0477 und EJ6788 aber es gibt 10 bis 20 mal soviel Schadensmeldungen auf dies Chargen. Das ist nur denkbar, wenn die beiden Chargen ganz schnell nach so vielen Toten vom Markt verschwand.
Welche StA geht diesen beiden Chargen einmal nach?
#EM0477 #EJ6788 #Todeschargen #Zurückgezogen #Skandal #Impschaden #Impfschäden #Impftote
Sehen wir uns die erste Charge EM0477 nach den Daten bei VEARS an:
234 Angehöriger und Ärzte meldeten nach Verabreichung von der Charge EM0477 den Tod nach dieser Impfung
148 Menschen meldeten dauerhafte Schwerbehinderung nach Verabreichung der Charge EM0477
109 Menschen deklarierten lebensbedrohliche Erkrankungen nach Verabreichung von EM0477
513 Ärzte meldeten Patienten nach Verabreichung der Charge EM0477, bei denen eine Behandlung im Krankenhaus erforderlich wurde
1.218 weitere Menschen meldeten weitere gesundheitliche Schäden nach Verabreichung on EM0477
und jetzt kommt es 79 Impfzentren und Ärzte sowie Angehörige meldeten sofort den Tod nach Verabreichung von EM0477. Das ist krass.
Sehen wir uns die nächst Charge an EJ6788
152 Angehöriger und Ärzte meldeten nach Verabreichung von der Charge EJ6788 den Tod nach dieser Impfung
38 Menschen meldeten dauerhafte Schwerbehinderung nach Verabreichung der Charge EJ6788
64 Menschen deklarierten lebensbedrohliche Erkrankungen nach Verabreichung von EJ6788
357 Ärzte meldeten Patienten nach Verabreichung der Charge EJ6788, bei denen eine Behandlung im Krankenhaus erforderlich wurde
651 weitere Menschen meldeten weitere gesundheitliche Schäden nach Verabreichung von EJ6788
und jetzt kommt es 30 Impfzentren, Ärzte und Angehörige meldeten sofort nach Verabreichung von EJ6788 den Tod dieser Menschen.
Wir glichen die beiden Chargen mit unserer Statistik ab. Die beiden vorstehenden Chargen übersteigen mindestens um das 5fach die Top 1 - Chargen in unserer eigenen Kanzlei Statistik, was Tote und Verletzte angeht. Für die erste Charge erklärten bisher aber nur 2 Mandanten, dass diese Charge schadenursächlich sei und für die 2 Charge EJ6788 nur 3 weitere Mandanten, dass diese Charge die Ursache für Impfschäden gesetzt habe. Die Statistik von VEARS und unsere Statistik divergiert gigantisch, weshalb es nur einen logischen Schluss gibt:
Die Charge EM0477 und die Charge EJ6788 wurden vom Markt durch Rückruf zurück genommen und zwar klammheimlich ohne irgendein Aufsehen zu erregen. Anders ist die Divergenz nicht erklärbar.
Anliegend in den Bildern sind die Top 5 aufgelistet. Jede dort angegebene Charge ist signifikant weniger schadensträchtig als EM0477 und EJ6788 aber es gibt 10 bis 20 mal soviel Schadensmeldungen auf dies Chargen. Das ist nur denkbar, wenn die beiden Chargen ganz schnell nach so vielen Toten vom Markt verschwand.
Welche StA geht diesen beiden Chargen einmal nach?
#EM0477 #EJ6788 #Todeschargen #Zurückgezogen #Skandal #Impschaden #Impfschäden #Impftote
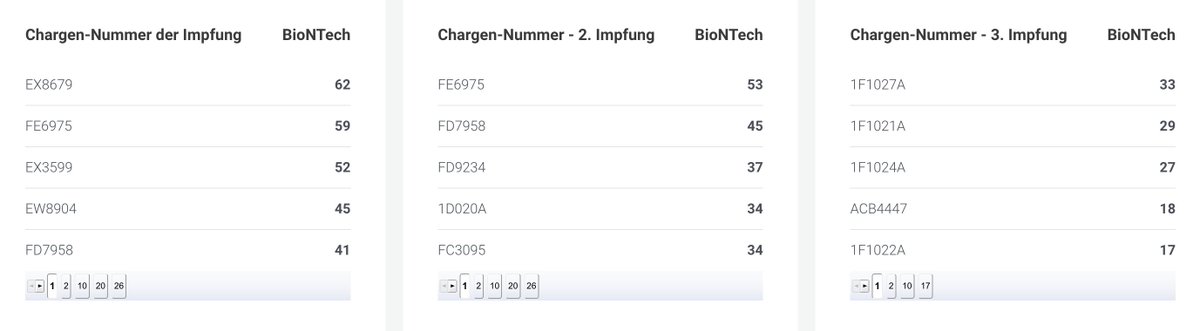
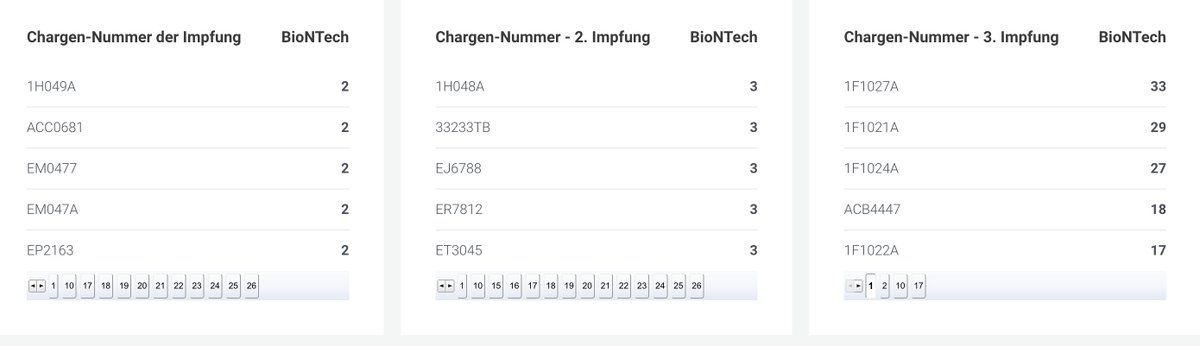


Hier noch einmal alle von Comirnaty erfassten 149 schadensträchtigen Chargen:
000086A 000106A 000112A 000114A 000124A 000125A 000128A 000130A 000136A 000137A 000139A 045G21A 092F21A
10020A 1C0008A 1C006A 1C007A 1C009A 1C010A 1CA008A 1D012A 1D013A 1D014A 1D015A 1D016A 1D017A 1D018A 1D020A 1DO13A 1DO15A 1E021A 1E026A 1E028A 1F030A 1F034A 1F036A 1F1004A 1F1010A 1F1021A 1F1022A 1F1024A 1F1027A 1F1042A 1F1049A 1F1052A 1G040A 1H049A
210004 210072 214008 214022 216044 3002537
3002620 3004233 3004500 3004951 3004954 30011TB 30025TB 30891TB 31043TB 31101TB 34523TB 33233TB
ABV3025 ABV3374 ABV4678 ABV5297 ABV5443 ABV6652 ABW4054 ABW4330 ABW7189 ACB3738 ACB4447 ACB4692 ACB4694 ACB5317 ACB5318 ACB8967 ACB9148 ACC0681 ACC1336
EJ6788 EJ6789 EJ6795 EJ6796 EK9788 EK9788 EM0477 EP9598 ER2659 ER7812 ER9480 ET3045 ET3025 ET3045 ET3674 EW8904 EX3510 EX3599 EX7823 EX8679 EX8680
FA5833 FC1440 FC3095 FC8885 FC8889 FD7958 FD9234 FE6975 FE7011 FE8405 FE9174 FF0900 FH9678 FN4072 FP1972
GE8310 GH9715
SCJU6 SCKT8 SCKX7 SCPT7 SCRM8 SCRP9 SCRW2 SCTD6 SCTJ2 SCTN4 SCUE1 SCUL2 SCVC6 SCVK4 SCVW7 SCVY8 SCWF3 SDEH4 SDEJ8 SDEW9 STVK4
XD955 XD974 XD975 XD985 XE395 XE423
000086A 000106A 000112A 000114A 000124A 000125A 000128A 000130A 000136A 000137A 000139A 045G21A 092F21A
10020A 1C0008A 1C006A 1C007A 1C009A 1C010A 1CA008A 1D012A 1D013A 1D014A 1D015A 1D016A 1D017A 1D018A 1D020A 1DO13A 1DO15A 1E021A 1E026A 1E028A 1F030A 1F034A 1F036A 1F1004A 1F1010A 1F1021A 1F1022A 1F1024A 1F1027A 1F1042A 1F1049A 1F1052A 1G040A 1H049A
210004 210072 214008 214022 216044 3002537
3002620 3004233 3004500 3004951 3004954 30011TB 30025TB 30891TB 31043TB 31101TB 34523TB 33233TB
ABV3025 ABV3374 ABV4678 ABV5297 ABV5443 ABV6652 ABW4054 ABW4330 ABW7189 ACB3738 ACB4447 ACB4692 ACB4694 ACB5317 ACB5318 ACB8967 ACB9148 ACC0681 ACC1336
EJ6788 EJ6789 EJ6795 EJ6796 EK9788 EK9788 EM0477 EP9598 ER2659 ER7812 ER9480 ET3045 ET3025 ET3045 ET3674 EW8904 EX3510 EX3599 EX7823 EX8679 EX8680
FA5833 FC1440 FC3095 FC8885 FC8889 FD7958 FD9234 FE6975 FE7011 FE8405 FE9174 FF0900 FH9678 FN4072 FP1972
GE8310 GH9715
SCJU6 SCKT8 SCKX7 SCPT7 SCRM8 SCRP9 SCRW2 SCTD6 SCTJ2 SCTN4 SCUE1 SCUL2 SCVC6 SCVK4 SCVW7 SCVY8 SCWF3 SDEH4 SDEJ8 SDEW9 STVK4
XD955 XD974 XD975 XD985 XE395 XE423
Herr @AlexWallasch hatte as PEI gefragt und eine Blitzerklärung erhalten, dass dem PEI keinerlei Kenntnis über die Unterschiede von Chargen vorliege. Das stimmt dann, wenn sie nach § 13 Abs. 5 IfSG nicht die Chargennummern zu den Verdachtsmeldungen auswerteten.
Verdachtsmeldungen seien alle irrelevant, diesen müsse die Pharmakovigilanzhehörde PEI nicht nachgehen. Knapp 400 Tote als Verdachtsmeldungen, die 10 Prozent aller deutschen Verdachtstoten in nur 2 Chargen ausmachten, hätten beim PEI, das selbst an die EMA und VEARS nach unsererm Kenntnisstand bis November 2021 die Daten lieferte, keine Veranlassung gesehen, im Rahmen der Arzneimittelaufsicht die Toten aufzuklären.
Die Rechtsanwältin, Frau Dr. Meyer - Hesselbarth, wollte die Auswertung des PEIs von den Chargen zu den Verdachtsmeldungen haben. Sie erklärten, dass zwar die Information als solche vorlägen, aber eine solche Auswertung nicht extra für das IFG - Verfahren anzufertigen sei, wenn die Auswertung nicht vorliege, womit sie erklärten, eine solche Auswertung nicht vorgenommen zu haben, weil sie ja ansonsten nach ihrer Diktion hätten eine solche vorlegen können. Das erklärt dann auch, wenn die Behörde gegen § 13 Abs. 5 Nr. 9 und 10 IfSG verstößt, dass ihr dann keinerlei Kenntnis vorliegt. Es war aber gesetzliche Pflicht des PEI.
In einer weiteren IFG - Anfrage von mir erklärte das PEI, dass sie die Anzahl der erworbenen und an die Ärzte und Impfzentren vertriebenen Chargen und Dosen nicht herausgeben würden, da es sich um ein Betriebsgeheimnis des Herstellers handele. Das betreffe auch die Zahl der Dosen pro Charge.
Es gibt dann zwei Schlussfolgerungen. Entweder sie haben in den IFG Anfragen gelogen und oder sie lügen jetzt, indem sie so tun, als hätten sie die Verdachtsmeldungen zu den Chargen ausgewertet.
Alle anderen Länder, Dänemark, Niederlande, Spanien, Tschechien und nun auch die USA erklären, dass nur 12 bis 15 Prozent der Chargen alle gesundheitlichen Schäden verursachten. Dann lügen all diese Länder und deren Gesundheitsministerien nach Angabe des PEI auch? Ferner ist es dann wohl nicht Aufgabe des PEI gewesen, den knapp 400 Todesmeldungen bei der Chargen von Seiten des PEI nachzugehen, erst recht, wenn diese nur bis April 2021 verimpft worden seien?
Die Bevölkerung sollte sich ernsthaft die Frage stellen, ob der Name "Arzneimittelaufsicht" überhaupt noch zutrifft oder ob es nicht eher ein Pharmalobbyblock der Impfhersteller ist, denen die Gesundheit der Bevölkerung am Allerwertesten vorbeigeht.
#Chargen #Todeschargen #Plazebos #Antwort #PEI
Verdachtsmeldungen seien alle irrelevant, diesen müsse die Pharmakovigilanzhehörde PEI nicht nachgehen. Knapp 400 Tote als Verdachtsmeldungen, die 10 Prozent aller deutschen Verdachtstoten in nur 2 Chargen ausmachten, hätten beim PEI, das selbst an die EMA und VEARS nach unsererm Kenntnisstand bis November 2021 die Daten lieferte, keine Veranlassung gesehen, im Rahmen der Arzneimittelaufsicht die Toten aufzuklären.
Die Rechtsanwältin, Frau Dr. Meyer - Hesselbarth, wollte die Auswertung des PEIs von den Chargen zu den Verdachtsmeldungen haben. Sie erklärten, dass zwar die Information als solche vorlägen, aber eine solche Auswertung nicht extra für das IFG - Verfahren anzufertigen sei, wenn die Auswertung nicht vorliege, womit sie erklärten, eine solche Auswertung nicht vorgenommen zu haben, weil sie ja ansonsten nach ihrer Diktion hätten eine solche vorlegen können. Das erklärt dann auch, wenn die Behörde gegen § 13 Abs. 5 Nr. 9 und 10 IfSG verstößt, dass ihr dann keinerlei Kenntnis vorliegt. Es war aber gesetzliche Pflicht des PEI.
In einer weiteren IFG - Anfrage von mir erklärte das PEI, dass sie die Anzahl der erworbenen und an die Ärzte und Impfzentren vertriebenen Chargen und Dosen nicht herausgeben würden, da es sich um ein Betriebsgeheimnis des Herstellers handele. Das betreffe auch die Zahl der Dosen pro Charge.
Es gibt dann zwei Schlussfolgerungen. Entweder sie haben in den IFG Anfragen gelogen und oder sie lügen jetzt, indem sie so tun, als hätten sie die Verdachtsmeldungen zu den Chargen ausgewertet.
Alle anderen Länder, Dänemark, Niederlande, Spanien, Tschechien und nun auch die USA erklären, dass nur 12 bis 15 Prozent der Chargen alle gesundheitlichen Schäden verursachten. Dann lügen all diese Länder und deren Gesundheitsministerien nach Angabe des PEI auch? Ferner ist es dann wohl nicht Aufgabe des PEI gewesen, den knapp 400 Todesmeldungen bei der Chargen von Seiten des PEI nachzugehen, erst recht, wenn diese nur bis April 2021 verimpft worden seien?
Die Bevölkerung sollte sich ernsthaft die Frage stellen, ob der Name "Arzneimittelaufsicht" überhaupt noch zutrifft oder ob es nicht eher ein Pharmalobbyblock der Impfhersteller ist, denen die Gesundheit der Bevölkerung am Allerwertesten vorbeigeht.
#Chargen #Todeschargen #Plazebos #Antwort #PEI


Eine inhaltliche Auseinandersetzung scheut auch der Gesundheitsminister, weshalb er mich als Organ der Rechtspflege und Anwalt der Impfgeschädigten geblockt hat. Gut zu wissen, auf wessen Seite er steht - nämlich der der Impfhersteller. Schließtlich zahlt er die Honorare deren US-Amerikanische für die Impfhersteller aus Steuergeldern und auch deren Gerichskosten und richtete dafür eine Koordinierungsstelle ein. Ein Gesundheitsminister der offen den Kampf gegen Impfgeschädigte aufnimmt und mit deren Interessenvertretern nicht kommuniziert ist in diesem Rechtsstaat untragbar.
#Lauterbach #blockt @AnwaltUlbrich
#Lauterbach #blockt @AnwaltUlbrich

• • •
Missing some Tweet in this thread? You can try to
force a refresh










