
Studium Berlin, Belgien, USA, GF
Rechtsanwälte, jur. spezialisiert auf Impfschäden



 Wer die Entscheidungsgründe alle lesen möchte. Ich stelle sie alle hintereinander Seite für Seite ein. Teil 2 Entscheidungsgründe
Wer die Entscheidungsgründe alle lesen möchte. Ich stelle sie alle hintereinander Seite für Seite ein. Teil 2 Entscheidungsgründe 





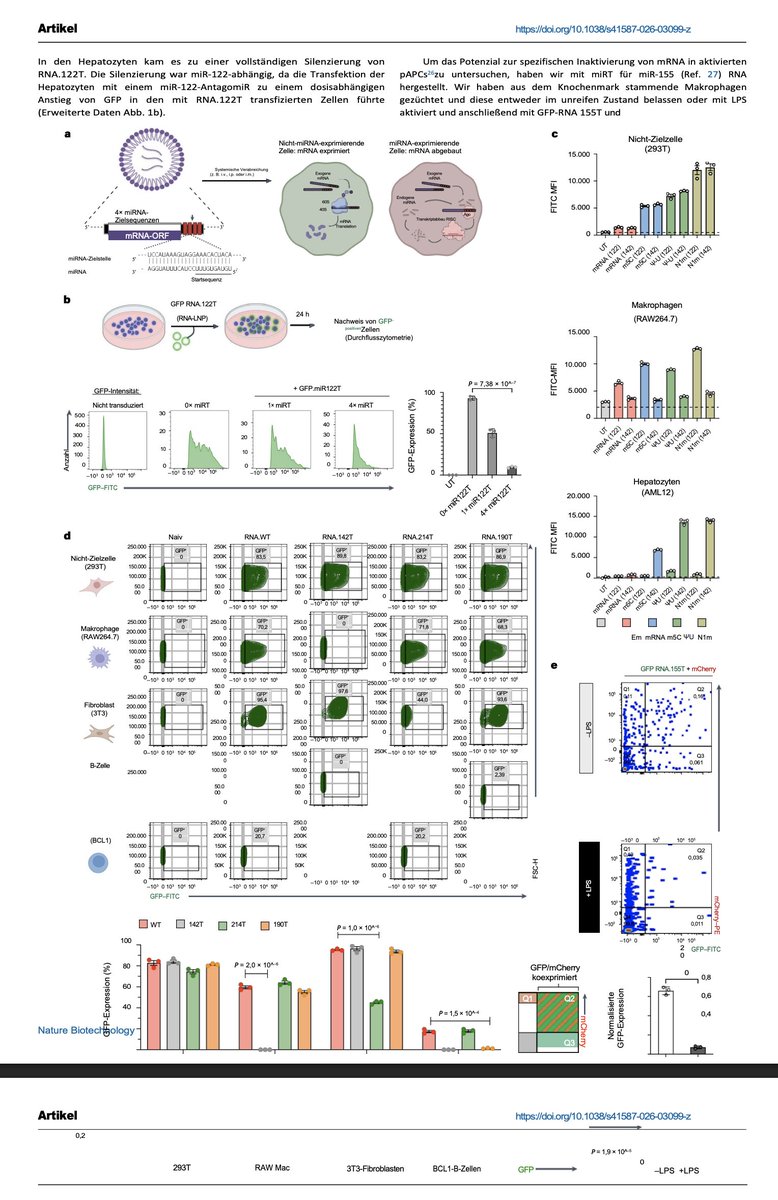




 Fortsetzung
Fortsetzung 



 Comirnaty wurde in OEB5 eingestuft. Hoch toxisch ab 1 Mikrogramm. Jeder kann ja mal im Internet heraussuchen, was das für den Arbeitsschutz heißt.
Comirnaty wurde in OEB5 eingestuft. Hoch toxisch ab 1 Mikrogramm. Jeder kann ja mal im Internet heraussuchen, was das für den Arbeitsschutz heißt. 
 Ach so- hier die Texte für den Schweizer Käse in Schimmelreinkultur jeweils Anhang I:
Ach so- hier die Texte für den Schweizer Käse in Schimmelreinkultur jeweils Anhang I: 


 Fortsetzung
Fortsetzung 






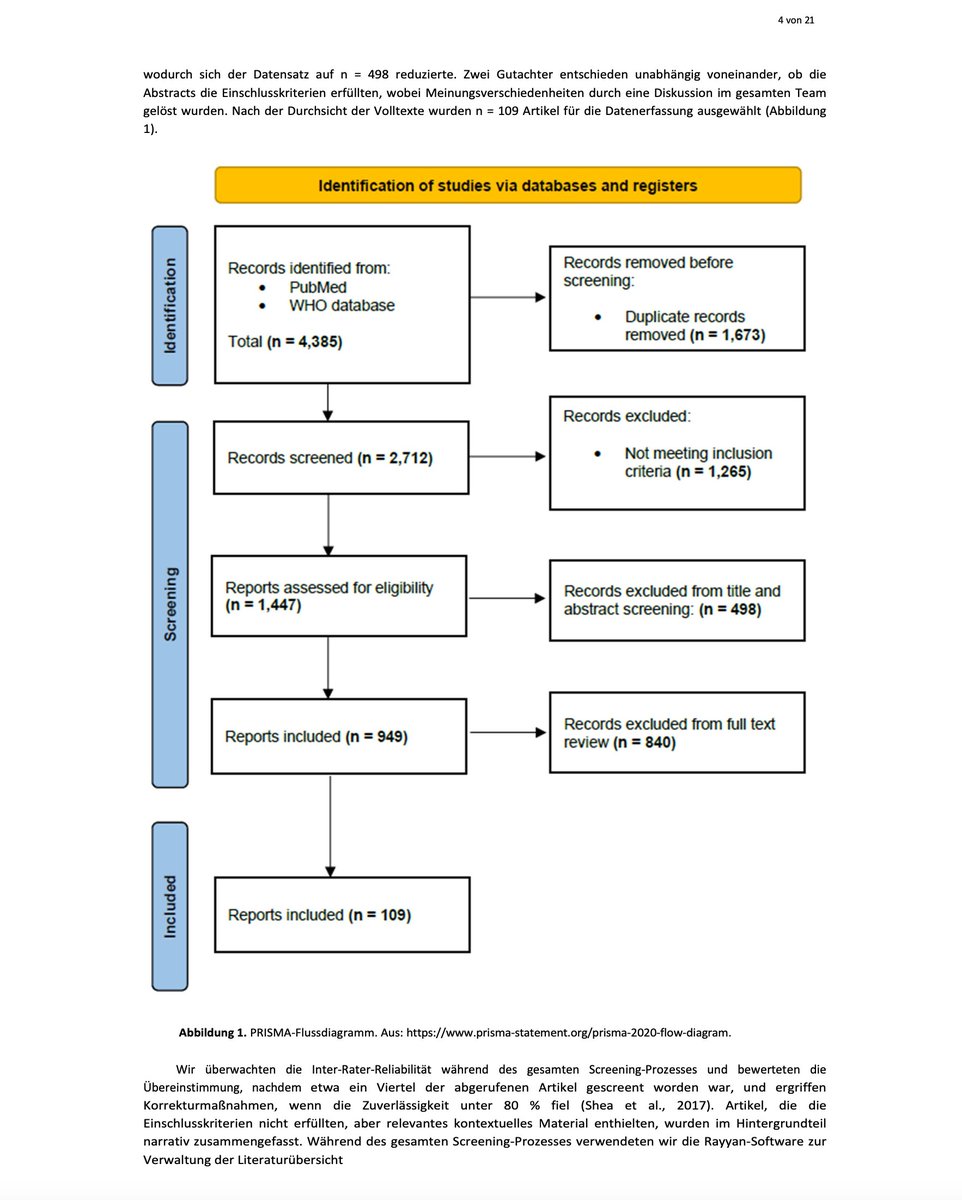 Fortsetzung
Fortsetzung 



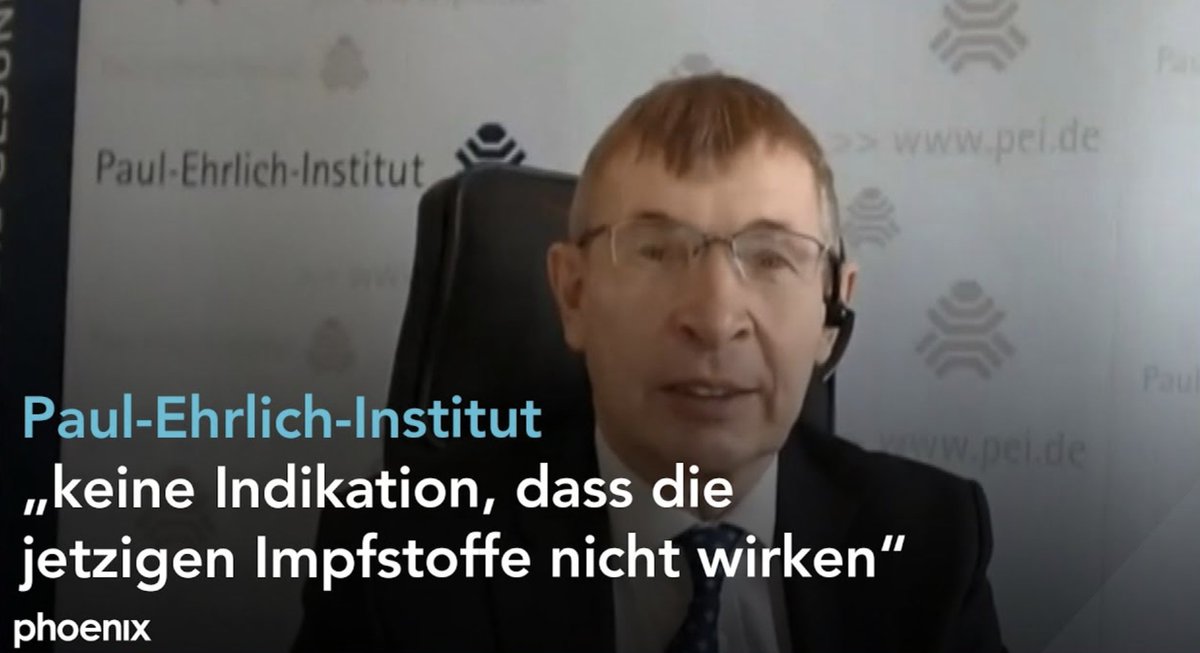
 laurakasner.substack.com/p/a-horrifying…
laurakasner.substack.com/p/a-horrifying…

 Vom PEI veröffentlichte schadensträchtige Chargen zu Verdachtsmeldungen von Moderna. Vorne die Chargennummer und anschließend die Anzahl der Verdachtsmeldungen, die beim PEI eingingen. Hier ist auch eine klare Chargenabhängigkeit zu erkennen.
Vom PEI veröffentlichte schadensträchtige Chargen zu Verdachtsmeldungen von Moderna. Vorne die Chargennummer und anschließend die Anzahl der Verdachtsmeldungen, die beim PEI eingingen. Hier ist auch eine klare Chargenabhängigkeit zu erkennen. 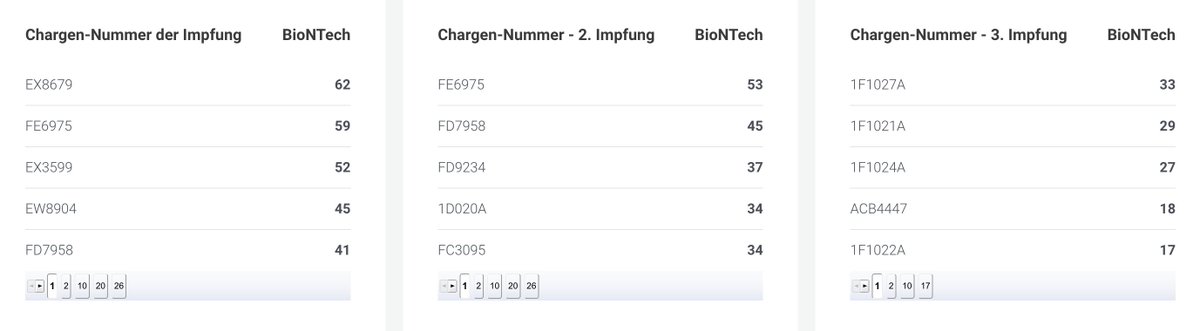
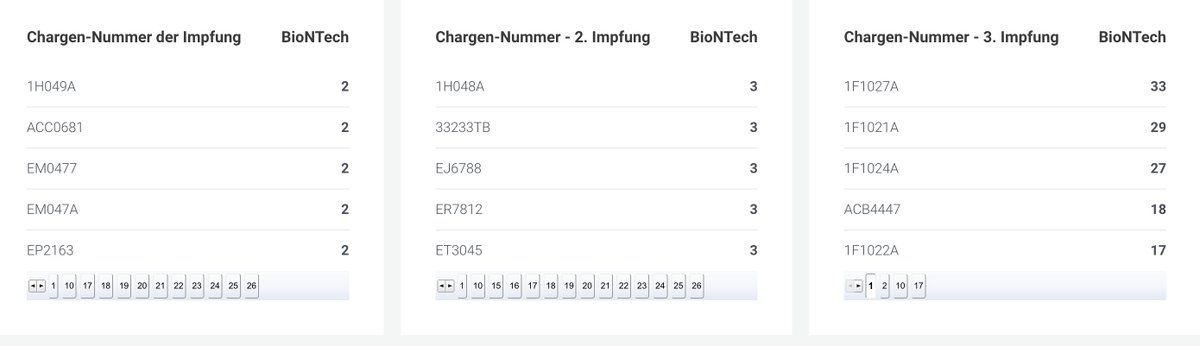

 Hier noch einmal alle von Comirnaty erfassten 149 schadensträchtigen Chargen:
Hier noch einmal alle von Comirnaty erfassten 149 schadensträchtigen Chargen:



 Ach so noch ganz wichtig!! Aus der bedingten Zulassung erwachse die Tatbestandswirkung und werde dann noch einmal durch die reguläre Zulassung bestätigt. In der Zulassung und den Anhängen finden sie keinen Satz zum Nutzen - Risiko - Verhältnis! Die freie richterliche Würdigung kommt dann zur vollen Tatbestandswirkung, während der ausdrücklichen Anerkennung als Impfschaden überhaupt keine Tatbestandswirkung zukommt.
Ach so noch ganz wichtig!! Aus der bedingten Zulassung erwachse die Tatbestandswirkung und werde dann noch einmal durch die reguläre Zulassung bestätigt. In der Zulassung und den Anhängen finden sie keinen Satz zum Nutzen - Risiko - Verhältnis! Die freie richterliche Würdigung kommt dann zur vollen Tatbestandswirkung, während der ausdrücklichen Anerkennung als Impfschaden überhaupt keine Tatbestandswirkung zukommt.  Anklageschrift Fortsetzung - Folge 2 - Nummerierung setzt im Original bei RN 25 weiter an. Twitter setzt nun neu an.
Anklageschrift Fortsetzung - Folge 2 - Nummerierung setzt im Original bei RN 25 weiter an. Twitter setzt nun neu an.