
BA.3.2 update: another sequence from the Netherlands, June 18 collection.
It belongs on the same branch as the GBW travel seq (tree gets confused by ORF7-8 deletion). Also, there are 3 artifactual muts in the GBW sequence (as usual), so the branch is shorter than it looks.
It belongs on the same branch as the GBW travel seq (tree gets confused by ORF7-8 deletion). Also, there are 3 artifactual muts in the GBW sequence (as usual), so the branch is shorter than it looks.
https://twitter.com/LongDesertTrain/status/1940384545741926911

Bottom line, in my view: BA.3.2 has spread internationally & is likely growing, but very slowly. If nothing changes, its advantage vs circulating lineages, which seem stuck in an evolutionary rut, will likely gradually grow as immunity to dominant variants solidifies... 2/9
So far, this seems like a slow-motion version of what we saw with BA.2.86, which spread internationally & grew very slowly for months. But then it got S:L455S & exploded, wiping out all competitors. Will something similar happen with BA.3.2? I think there's a good chance... 3/9

BA.2.86 had a glaring weakness in its vulnerability to a particular class of type-1 antibodies. @SolidEvidence predicted (almost) exactly what would happen: BA.2.86 would get a mutation at 455-456 & take off. Shortly thereafter, it happened. 4/
https://x.com/SolidEvidence/status/1704570301634461876
BA.3.2 has two weaknesses.
First, it remains vulnerable to type-1 antibodies that LF.7.9 & XFG have managed to escape—a key advantage they have over the formerly dominant LP.8.1.1.
If it can find a way to caulk this chink in its armor, BA.3.2 may be able to pull a JN.1. 5/
First, it remains vulnerable to type-1 antibodies that LF.7.9 & XFG have managed to escape—a key advantage they have over the formerly dominant LP.8.1.1.
If it can find a way to caulk this chink in its armor, BA.3.2 may be able to pull a JN.1. 5/
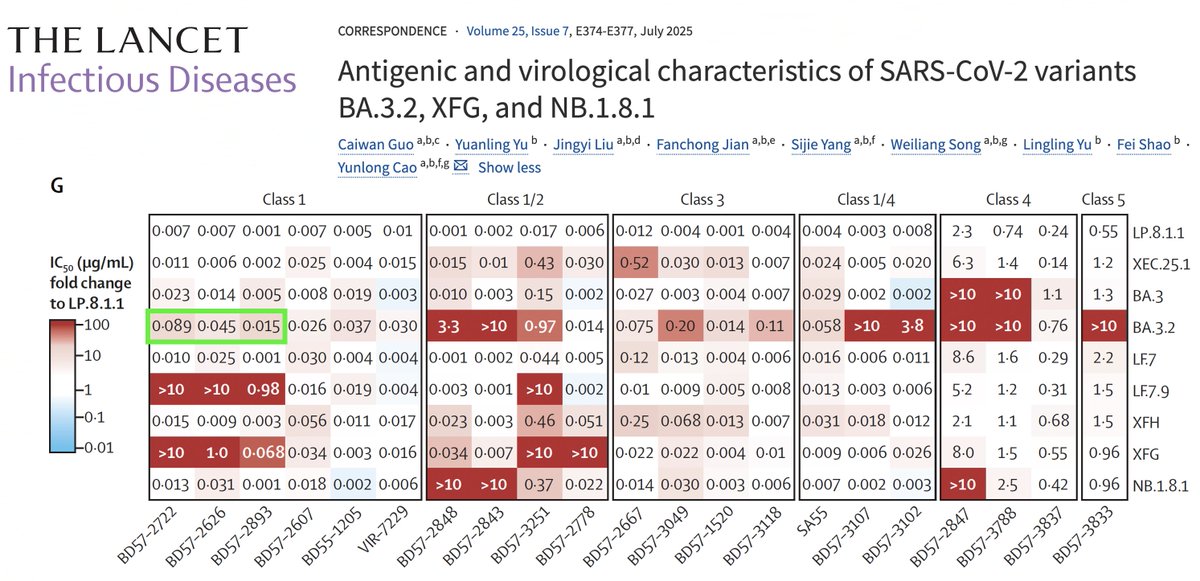
Given the distinguishing LF.7.9 & XFG muts, they most likely are able to dodge these type-1 antibodies via spike mutations:
• L441R + K444R + H445P + A475V (LF.7.9)
• K444R + H445R + N487D (XFG)
There are likely many other mutations that could do the job as well. 6/9
• L441R + K444R + H445P + A475V (LF.7.9)
• K444R + H445R + N487D (XFG)
There are likely many other mutations that could do the job as well. 6/9
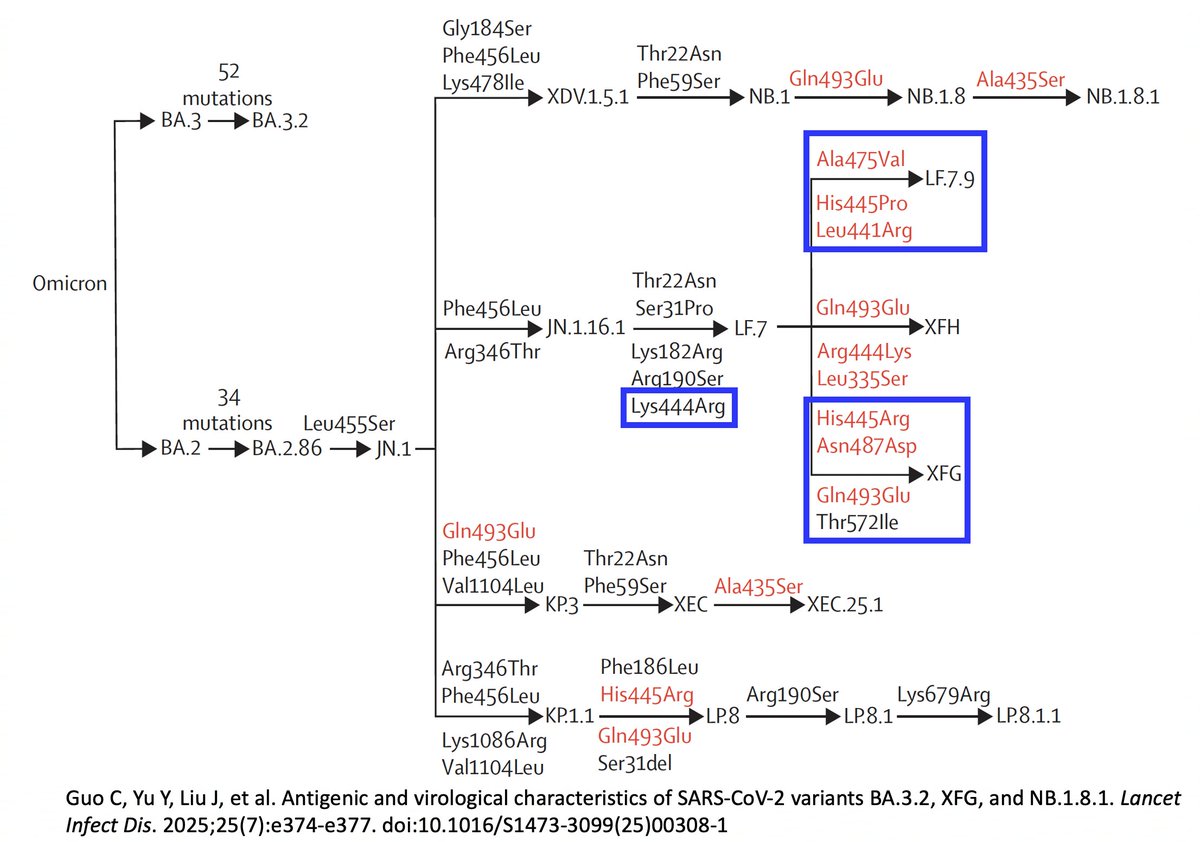
The other major BA.3.2 flaw is its weak binding to ACE2 the cell receptor the virus uses to enter cells.
In this sense, it is similar to XBB.1, which was also the most immune-evasive variant around, but which grew very slowly because of its weak ACE2 affinity. 7/9
In this sense, it is similar to XBB.1, which was also the most immune-evasive variant around, but which grew very slowly because of its weak ACE2 affinity. 7/9

Then XBB.1 got S486P, which conferred a huge bump in ACE2 binding, and the game was up. XBB's dominated for the next 12 months—when JN.1 arrived. 8/9

The BA.3.2 spike is different from any we've seen, so we don't know what mutations are viable on its background.
But I have to believe there's a viable path to erasing its weaknesses. The longer & more widely it circulates, the greater chance it has of finding the solution.
9/9
But I have to believe there's a viable path to erasing its weaknesses. The longer & more widely it circulates, the greater chance it has of finding the solution.
9/9
• • •
Missing some Tweet in this thread? You can try to
force a refresh










