
🚨New paper alert!🚨 Cells integrate signals into decision-making. But how do levels of signaling influence cell-fate transitions? Is more always better? Or is there an “optimal” level for cell-fate programming? If we could tune signaling, could optimize production of neurons? Read on! (1/n)🧵

To ask this question, Brittany started probing the the MAPK pathway. The MAPK pathway is a central regulator of cellular decision-making. The levels and dynamics of signaling direct cells down different fate. Based on previous work, we knew that adding a MAPK mutant (HRAS-G12V) increases direct conversion of skin cells to motor neuron when combined motor neuron TFs (NIL) and a p53 mutant. (2/n)

Adding of HRAS-G12V increase proliferation, expanding the number of cells that can convert. But also, hyperproliferative cells are more likely (on a per-cell basis) to convert and they adopt more neuronal features. (3/n)

The canonical phosphorylation cascade transmits extracellular signals (growth factors, etc) to drive changes in gene expression, which lead to proliferation, differentiation. (4/n)

Overexpression of mutant RAS drives higher signaling rates which increase proliferation. Many cancers can be tracked to mutants in RAS and other MAPK components. Sitting upstream, Ras is very effective in driving signaling changes into cell-fate changes. But…sometime RAS doesn’t increase cell-fate transitions…why? (5/n)

We wondered if the exact level of RAS expression might influence cell-fate transitions. So Brittany decided to titrate HRAS a combination of approaches including lenti dose, transgene design, etc. Using a twin assay approach, she could measure HRAS-mCherry levels early in conversion and then measure the resulting rates of conversion. And then…(6/n)

What she saw was amazing! Indeed, when controlling for batch variation, she could see this beautiful biphasic curve! 😍 Finding optimums in biological systems is extremely challenging but Brittany figured out how to capture this biphasic response! (7/n)

But she wondered if her results would be useful for designing optimal cassettes of expression. She hypothesized that cassettes of our cocktail that had medium levels of expression of HRas would give the highest levels of conversion while extremes on either end would do poorly. So she built these cassettes with p53DD around an IRES and tested the rates of conversion (8/n)

Again, she saw a range of HRAS levels and a corresponding biphasic conversion response! The optimal cassette had medium levels of HRAS expression as she had predicted (9/n)

She wondered why high levels of HRAS were reducing conversion rates. HRAS overexpression can trigger oncogene-induced senescence (10/n)

By staining for B-gal, she saw increases in senescent cells and “fried egg” morphology. (11/n)

Senescence correlated with high levels of HRAS expression and lower rates of conversion to induced motor neurons. But was it the levels of expression of HRAS or the signaling they induced that led to the senescent state? (12/n)

Brittany wondered if she could improve conversion rates by “tuning down” signal using small molecule that inhibits MAPK signaling. (12/n)

And yes, indeed, she could reduce MAPK signaling and reduce senescence with the inhibitor! And indeed, this rescued conversion rates even when cells expressed HRAS at high levels. So it is the signaling level that needs tuning to an optimal level! 🙌 (13/n)

So now that we could tune down levels with a small molecule, could we tune them up? Could we replace HRAS with a small-molecule activator of MAPK signaling (PMA)? Or did we really need to overexpress HRAS? (14/n)

Amazingly, addition of PMA could replace HRAS and support high rates of conversion! (15/n)

And if we reduce signaling via MEKi, this effect is lost! Again, demonstrating that the optimal levels of signaling are what drive high rates of conversion. (16/n)

So from this work we see that optimal levels of MAPK signaling drive cells to a receptive states that supports high rates of conversion, whereas high signaling trigger senescence, explaining the biphasic response. Thus, by tuning the levels HRAS or pathway activity we could increase or decrease conversion. (17/n)
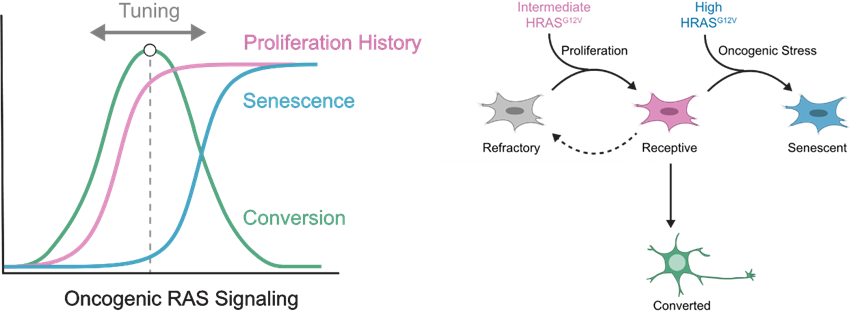
But we wondered, if we are using neuronal transcription factors, how are these TFs not blocking proliferation? How are cells able to proliferate and gain access to this receptive state? Could MAPK signaling be transiently affecting TFs like Ngn2? (18/n)

Previous work indicated that TFs like Ngn2 could be phosphorylated via the MAPK called ERK. Phosphorylation changed their properties such as binding and stability. Indeed, phospho-mutants of Ngn2 showed greater potency in generating neurons from progenitor cells. (19/n)

So we wondered what would happen to conversion rates if we overexpressed the phosphomutant/ Overall, our yields went down. The phospho-mutant reduced proliferation so there were fewer neurons. But the mutant seemed more potent with less receptive cells (20/n)

So we wondered what would happen to the levels of Ngn2 with MAPK signaling active or inhibited? Could the phosphorylation reduce Ngn2 levels of the WT? (21/n)

Indeed, when the MAPK pathway is “ON” we see very little Ngn2. In particular, the top band in the Western blot gone. When MAPK signaling is low or OFF, we see different states of Ngn2 which likely reflect phosphorylation (22/n).

So we updated our model a bit to show that MAPK specifically influences Ngn2 levels, allowing cells to proliferate by reducing Ngn2 (transiently!), paving the way for induction to neurons later in conversion. (23/n)
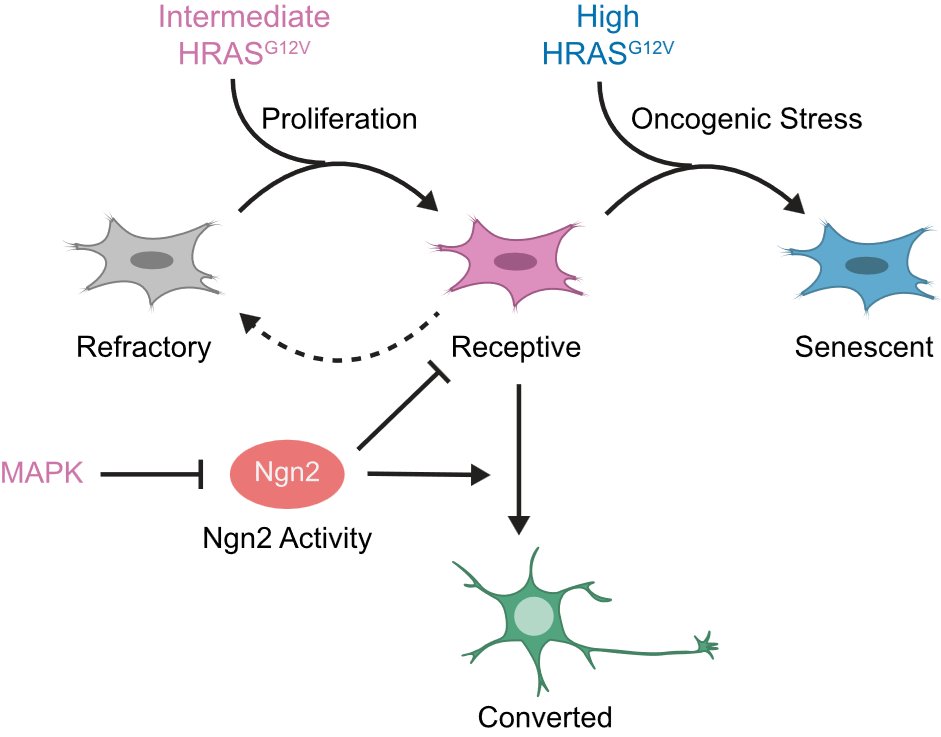
Overall, we find: 1) Cell-fate programming responds biphasically to HRASG12V expression 2) A small-molecule MAPK inducer can replace HRASG12V for high rates of conversion 3) MAPK signaling alters Ngn2 levels, influencing proliferation and conversion (24/n)

🎯 Other take-homes: An optimal 'just-right' MAPK level drives efficient, safe, high-yield conversion. This tuning framework could generalize to other fate transitions. Also, we see that the presence and levels of other oncogenes tunes the exact response to HRAS, adding further nuance to how different signals are processed by cells into cell-fate transitions. (25/n)
🚀 Why it matters: Here we uncover a quantitative 'map' of how MAPK signaling shapes identity which offers a new lever for design of therapies for regenerative medicine and demonstrates how chemical tuning can replace oncogenic drivers safely, but there’s more to explore in this space on how chemical tuning influences neuronal features (26/n)
👩🔬 This project was expertly by Brittany Lende-Dorn who dug into the quantitative and wet lab challenges with enormous focus and chased the data to build a great story. Joining her were two of our UGs with Jane Atkinson & Yunbeen Bae who learned direct conversion (not trivial) and were able to independently contribute to the story! Huge congrats to everyone! (27/n)

💵We are so very grateful for the wonderful team at Cell Reports and the support from NIH, NSF, and the Army’s Institute for Collaborative Biotechnologies. Without this foundational support, this work (and many other projects in the lab) could not have moved forward as far and as fast! So thank you! (28/n)
We’re excited to explore precise, tunable control of signaling for regenerative & therapeutic reprogramming. Also, not the cover for the issue, but Brittany illustrated this amazing vision of tuning MAPK signaling to optimize neuron production. Hope you enjoy! (29/n)

Loved the tweetorial and want to read the paper? See here: 🧠 'Chemogenetic tuning reveals optimal MAPK signaling for cell-fate programming.' And stay tuned for how we used these insights with a new tool that vastly simplifies titrations… Coming soon… Fin!doi.org/10.1016/j.celr…
@threadreaderapp unroll
• • •
Missing some Tweet in this thread? You can try to
force a refresh















