Can you take a quarter cup of composite sewage, simply ask ‘what’s in there?’, and find out all of the pathogens circulating in that community?
That is the question we asked in our latest pre-print.
Turns out you can.
1/
medrxiv.org/content/10.110…
That is the question we asked in our latest pre-print.
Turns out you can.
1/
medrxiv.org/content/10.110…
We are not the first group to do unbiased sequencing of wastewater to monitor circulating viruses, but I think we are the first to ever do it at this scale.
Weekly wastewater samples for 18 months, totaling over 85 Billion sequence reads.
2/
Weekly wastewater samples for 18 months, totaling over 85 Billion sequence reads.
2/

Among the ‘known’ viruses, there was a fairly even split between bacteria viruses (phages) and eukaryotic viruses.
This was just raw reads though, if you look at diversity there was considerably more species of phages.
3/
This was just raw reads though, if you look at diversity there was considerably more species of phages.
3/

Focusing on the eukaryotic viruses, you see that the vast majority of the eukaryotic viruses are Virgaviridae, which infect plants.
4/
4/

What plant viruses you may ask? The most prominent one is a virus called Tomato Brown Rugose Fruit Virus (ToBRFV).
This was true everywhere in the country.
Americans eat a lot of tomatoes.
5/
This was true everywhere in the country.
Americans eat a lot of tomatoes.
5/

What’s sort of surprising is that ToBRFV isn’t even in US tomatoes.
That explains why we see it year round though. These are probably from imported tomatoes and tomato products.
6/
en.wikipedia.org/wiki/Tomato_br…
That explains why we see it year round though. These are probably from imported tomatoes and tomato products.
6/
en.wikipedia.org/wiki/Tomato_br…
The one time of year when the ToBRV proportion goes down is late Summer, when it is partially displaced by Tomato Mosaic virus (which does infect US tomatoes).
This probably reflects people eating more local tomatoes when they are in season.
7/
This probably reflects people eating more local tomatoes when they are in season.
7/

Although human pathogens were a tiny fraction of the total sequences, there was still plenty of sequences to figure out what the circulating human pathogens were.
8/
8/

There was only one respiratory virus that was present year-round. You guessed it, SARS-CoV-2.
It’s still here.
9/
It’s still here.
9/

We were also monitoring SARS-CoV-2 in these samples the old-fashioned way (dPCR) and it was nice to see that the amount detected from sequencing (normalized or not normalized) correlated pretty well with the dPCR results.
10/
10/

We also detected all of the other human coronaviruses, and influenza viruses. They all were most prevalent January-March, as expected.
10/
10/

Other respiratory viruses circulated later. For instance, ParaInfluenza 3 circulated from April-June both years.
This was expected, but I still don’t understand it epidemiologically.
Why then?
11/
This was expected, but I still don’t understand it epidemiologically.
Why then?
11/

Most of the Rhinoviruses also circulated in the Spring, but as I’ve noted before, the specific serotypes changed from year to year.
12/
12/

You can see a much more detailed readout of the rhinovirus (with more sites and dates) on our dashboard.
13/
dholab.github.io/public_viz/004…
13/
dholab.github.io/public_viz/004…
We also saw a year-to-year turnover in Parechovirus serotypes (causes meningitis).
This exactly matched what our colleagues down the road in KC found in pediatric patients.
14/ pubmed.ncbi.nlm.nih.gov/40712199/
This exactly matched what our colleagues down the road in KC found in pediatric patients.
14/ pubmed.ncbi.nlm.nih.gov/40712199/
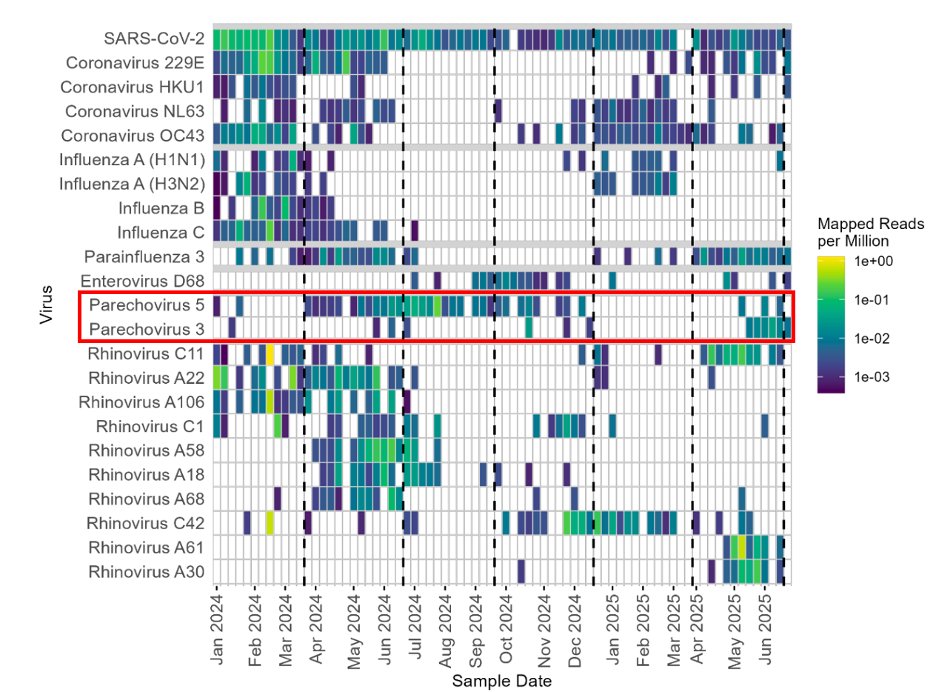
There were also a few Fall respiratory viruses we detected. Enterovirus D68 (expected), and on off-season surge of rhinovirus C42 (not expected, but nationwide).
15/
15/

There were other things we saw that we REALLY didn’t expect. For instance, influenza H5N1 B3.13 (which wasn't in MO) appeared in Spring of 2024.
We’re pretty sure this came from a dairy in town that imports their milk from Texas, where H5N1 was rampant at that time.
16/
We’re pretty sure this came from a dairy in town that imports their milk from Texas, where H5N1 was rampant at that time.
16/

The data in this paper is from one sewershed Jan.2024-June 2025.
If you want to know about more sites and more recent data, visit our dashboard. It’s updated at least once a week.
17/
lungfish-science.github.io/wastewater-das…
If you want to know about more sites and more recent data, visit our dashboard. It’s updated at least once a week.
17/
lungfish-science.github.io/wastewater-das…
I think this kind of surveillance is the way of the future, but we’re not there yet.
1. Price: too expensive.
2. Unknowns: most sequences are 'unknown'.
3. False positives: requires careful curation.
18/
1. Price: too expensive.
2. Unknowns: most sequences are 'unknown'.
3. False positives: requires careful curation.
18/
Price, right now the sequencing alone is at least ~$500-1000 per sample.
However, it keeps going down. There was a noticeable decrease in price even during the duration of this study.
19/
However, it keeps going down. There was a noticeable decrease in price even during the duration of this study.
19/
Unknowns. A very large portion of the sequence from this data is ‘dark matter’: sequences from species (largely viruses) that have never been characterized.
(That’s what our next manuscript will be about.)
20/
(That’s what our next manuscript will be about.)
20/
False positives. Even many human viruses are not characterized.
Every week we find sequences whose closest match is polio, but it's never polio.
It’s always viruses related to polio that just aren’t in the database.
It's annoying, and time consuming checking them all.
21/
Every week we find sequences whose closest match is polio, but it's never polio.
It’s always viruses related to polio that just aren’t in the database.
It's annoying, and time consuming checking them all.
21/
Eventually sequencing will be cheaper, databases will be more complete, and this kind of study will be routine (maybe 5-10 years).
Meanwhile, keep pooping, we take this shit seriously.
Thanks to our collaborators/funders:
Inkfish
@SecureBio
22/22naobservatory.org/casper/
Meanwhile, keep pooping, we take this shit seriously.
Thanks to our collaborators/funders:
Inkfish
@SecureBio
22/22naobservatory.org/casper/
• • •
Missing some Tweet in this thread? You can try to
force a refresh