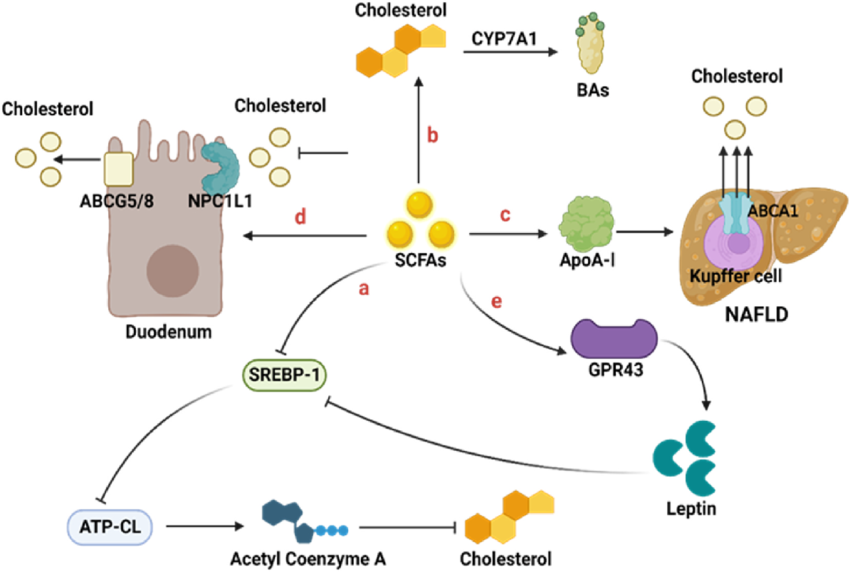
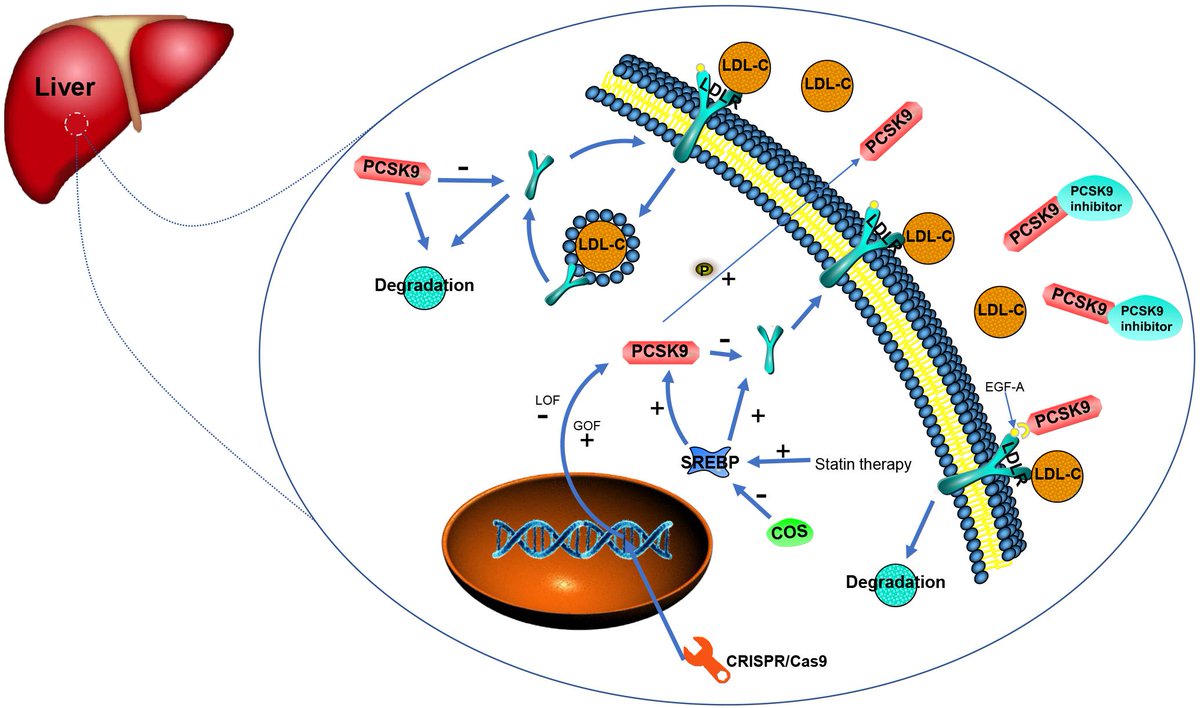
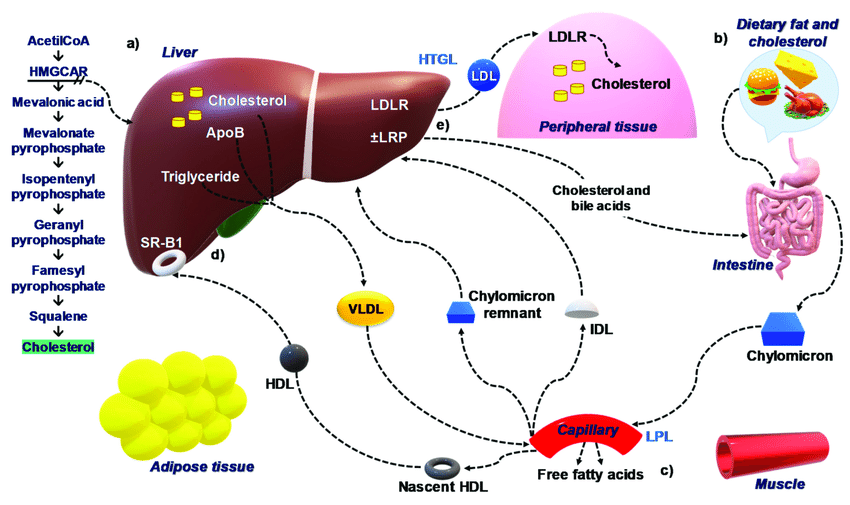
An extremely neglected aspect of hair growth/slowing down hair loss is monitoring inflammation.
Here's what you need to know.
Thread🧵
Here's what you need to know.
Thread🧵



It's George.
Monitoring inflammation is usually neglected because hair loss is traditionally treated only as a hormonal or genetic issue.
Follicle inflammation happens deep under the skin and often develops silently without visible surface signs.
But this chronic condition directly attacks hair stem cells, permanently stopping the creation of new strands.
It also causes micro-scarring around the root, choking out the vital nutrients the follicle needs.
Furthermore, the body diverts its cellular energy toward fighting inflammation instead of fueling new hair growth.
Ultimately, internal inflammation acts as an accelerator, triggering hereditary pattern baldness much earlier in life.
Monitoring inflammation is usually neglected because hair loss is traditionally treated only as a hormonal or genetic issue.
Follicle inflammation happens deep under the skin and often develops silently without visible surface signs.
But this chronic condition directly attacks hair stem cells, permanently stopping the creation of new strands.
It also causes micro-scarring around the root, choking out the vital nutrients the follicle needs.
Furthermore, the body diverts its cellular energy toward fighting inflammation instead of fueling new hair growth.
Ultimately, internal inflammation acts as an accelerator, triggering hereditary pattern baldness much earlier in life.
While inflammation is essential for survival and one of the most fundamental processes in the human body, it can also become harmful when excessive, prolonged or misdirected, leading to tissue damage, fibrosis, chronic disease and so on.
That’s why if you have a chronic health issue that you are trying to resolve, it’s very unlikely that you haven’t stumbled across terms such as “anti-inflammatories”, “inflammation cascade” and so on.
But what is inflammation exactly?
At its core, as you will find in most of its definitions, inflammation is the body’s protective response to injury or infection in vascularized living tissue.
So it occurs only in tissues with blood vessels and involves both vascular and cellular components.
It is broadly divided into acute and chronic forms.
Acute inflammation is dominated by neutrophils, begins within minutes to hours and lasts hours to days.
Examples include bacterial pneumonia, appendicitis or a simple cutaneous infection.
Chronic inflammation, in contrast, develops slowly and persists for weeks, months or years.
Rheumatoid arthritis, Crohn’s disease and atherosclerosis are some of the classic examples.
That’s why if you have a chronic health issue that you are trying to resolve, it’s very unlikely that you haven’t stumbled across terms such as “anti-inflammatories”, “inflammation cascade” and so on.
But what is inflammation exactly?
At its core, as you will find in most of its definitions, inflammation is the body’s protective response to injury or infection in vascularized living tissue.
So it occurs only in tissues with blood vessels and involves both vascular and cellular components.
It is broadly divided into acute and chronic forms.
Acute inflammation is dominated by neutrophils, begins within minutes to hours and lasts hours to days.
Examples include bacterial pneumonia, appendicitis or a simple cutaneous infection.
Chronic inflammation, in contrast, develops slowly and persists for weeks, months or years.
Rheumatoid arthritis, Crohn’s disease and atherosclerosis are some of the classic examples.


What can trigger inflammation?
Virtually any insult capable of causing tissue damage can initiate inflammation, whether that’s high blood sugar, working out, sunlight exposure, mold, cortisol, a virus or a burn.
Now in order to understand the acute inflammatory response, we must understand that it unfolds through three interconnected components:
-Vascular changes
-Cellular recruitment
-Mediator release
Vascular changes begin with transient vasoconstriction lasting seconds, followed by sustained vasodilation mediated by histamine and prostaglandins.
This increases blood flow, producing redness and warmth.
Simultaneously, vascular permeability rises through several mechanisms that are triggered by histamine and leukotrienes, for example.
The resulting leakage of protein-rich fluid forms an exudate(*), contributing to swelling.
Cellular recruitment follows a highly ordered sequence known as leukocyte extravasation.
First, leukocytes marginate to the vessel periphery and roll along the endothelium via reversible interactions between selectins and their ligands.
Chemokines then activate leukocyte integrins, which bind firmly to endothelial adhesion molecules such as ICAM-1 and VCAM-1.
The leukocyte then transmigrates between endothelial cells using PECAM-1, migrates toward the injury site along a chemokine gradient, and finally phagocytoses the target after opsonization by IgG or complement fragment C3b.
Finally, chemical mediators derive from plasma or cells.
Plasma systems include the complement cascade, generating anaphylatoxins C3a and C5a, the kinin system, producing bradykinin and the coagulation cascade.
Cell-derived mediators include histamine from mast cells and basophils, causing early vasodilation, arachidonic acid metabolites such as prostaglandins and leukotrienes and cytokines from macrophages and other cells.
(*)The fluid that leaks into tissues during inflammation is called an exudate.
Virtually any insult capable of causing tissue damage can initiate inflammation, whether that’s high blood sugar, working out, sunlight exposure, mold, cortisol, a virus or a burn.
Now in order to understand the acute inflammatory response, we must understand that it unfolds through three interconnected components:
-Vascular changes
-Cellular recruitment
-Mediator release
Vascular changes begin with transient vasoconstriction lasting seconds, followed by sustained vasodilation mediated by histamine and prostaglandins.
This increases blood flow, producing redness and warmth.
Simultaneously, vascular permeability rises through several mechanisms that are triggered by histamine and leukotrienes, for example.
The resulting leakage of protein-rich fluid forms an exudate(*), contributing to swelling.
Cellular recruitment follows a highly ordered sequence known as leukocyte extravasation.
First, leukocytes marginate to the vessel periphery and roll along the endothelium via reversible interactions between selectins and their ligands.
Chemokines then activate leukocyte integrins, which bind firmly to endothelial adhesion molecules such as ICAM-1 and VCAM-1.
The leukocyte then transmigrates between endothelial cells using PECAM-1, migrates toward the injury site along a chemokine gradient, and finally phagocytoses the target after opsonization by IgG or complement fragment C3b.
Finally, chemical mediators derive from plasma or cells.
Plasma systems include the complement cascade, generating anaphylatoxins C3a and C5a, the kinin system, producing bradykinin and the coagulation cascade.
Cell-derived mediators include histamine from mast cells and basophils, causing early vasodilation, arachidonic acid metabolites such as prostaglandins and leukotrienes and cytokines from macrophages and other cells.
(*)The fluid that leaks into tissues during inflammation is called an exudate.
Its composition varies with the insult.
Serous exudates are watery with low protein content, as seen in blisters or early pleural effusions.
Fibrinous exudates are rich in fibrinogen that polymerizes into fibrin.
Purulent or suppurative exudates contain neutrophils, necrotic debris, and microorganisms, forming pus in abscesses or bacterial pneumonias.
Hemorrhagic exudates contain red blood cells and occur in viral infections or malignancies.
Pseudomembranous exudates combine fibrin and necrotic epithelium, as in Clostridium difficile colitis.
Serous exudates are watery with low protein content, as seen in blisters or early pleural effusions.
Fibrinous exudates are rich in fibrinogen that polymerizes into fibrin.
Purulent or suppurative exudates contain neutrophils, necrotic debris, and microorganisms, forming pus in abscesses or bacterial pneumonias.
Hemorrhagic exudates contain red blood cells and occur in viral infections or malignancies.
Pseudomembranous exudates combine fibrin and necrotic epithelium, as in Clostridium difficile colitis.
Now, when tissue damage is minimal and the stimulus is efficiently cleared, complete resolution occurs with restoration of normal architecture, as in resolving lobar pneumonia.
But if damage is substantial, healing proceeds by fibrosis and scar formation.
Suppurative inflammation may wall off into an abscess. If the inciting agent persists or the response is dysregulated, acute inflammation transitions to a chronic phase.
Now chronic inflammation is characterized by persistent mononuclear cell infiltration, ongoing tissue destruction, and attempted repair through angiogenesis and fibrosis.
Granulomatous inflammation represents a specialized chronic response in which macrophages transform into epithelioid cells and fuse into multinucleated giants. Here, inflammation extends beyond local tissues. Fever results from macrophage-derived IL-1 and TNF-alpha inducing prostaglandin E2 in the hypothalamus.
The liver mounts an acute-phase response under IL-6 signaling, increasing C-reactive protein, fibrinogen, and serum amyloid A while decreasing albumin. Leukocytosis occurs with neutrophilia in bacterial infections and lymphocytosis in viral ones.
Prolonged TNF-alpha elevation causes cachexia through appetite suppression and muscle wasting.
If these were too complicated, think that inflammation is the immune system in action.
It is not a byproduct, a side effect or a separate process but the integrated, coordinated response of innate and adaptive immune cells, soluble mediators and non-immune tissues (endothelium, fibroblasts, adipocytes) that attempt to restore homeostasis.
That's why when adaptive immunity goes rogue (think Th17 in psoriasis, Th1 in TB granuloma) there's always chronic inflammation.
But if damage is substantial, healing proceeds by fibrosis and scar formation.
Suppurative inflammation may wall off into an abscess. If the inciting agent persists or the response is dysregulated, acute inflammation transitions to a chronic phase.
Now chronic inflammation is characterized by persistent mononuclear cell infiltration, ongoing tissue destruction, and attempted repair through angiogenesis and fibrosis.
Granulomatous inflammation represents a specialized chronic response in which macrophages transform into epithelioid cells and fuse into multinucleated giants. Here, inflammation extends beyond local tissues. Fever results from macrophage-derived IL-1 and TNF-alpha inducing prostaglandin E2 in the hypothalamus.
The liver mounts an acute-phase response under IL-6 signaling, increasing C-reactive protein, fibrinogen, and serum amyloid A while decreasing albumin. Leukocytosis occurs with neutrophilia in bacterial infections and lymphocytosis in viral ones.
Prolonged TNF-alpha elevation causes cachexia through appetite suppression and muscle wasting.
If these were too complicated, think that inflammation is the immune system in action.
It is not a byproduct, a side effect or a separate process but the integrated, coordinated response of innate and adaptive immune cells, soluble mediators and non-immune tissues (endothelium, fibroblasts, adipocytes) that attempt to restore homeostasis.
That's why when adaptive immunity goes rogue (think Th17 in psoriasis, Th1 in TB granuloma) there's always chronic inflammation.
Now let’s move on to the mediators of inflammation (these molecules are not just relevant to inflammation).
These are: cytokines, chemokines, lipids, ROS / RNS, proteases and histamine.
So let’s break each one down now.
First we have pro-inflammatory cytokines.
These are small secreted proteins that amplify and coordinate inflammation.
The main ones are TNF-alpha, IL-1, IL-6, and IL-8.
TNF-alpha, is mainly secreted by (activated) macrophages and Th1 cells in response to bacterial lipopolysaccharide, exists as a transmembrane precursor cleaved by the enzyme TACE into a soluble trimer.
It plays key roles in the pathogenesis of autoimmune diseases such as rheumatoid arthritis (RA), IBD and psoriatic arthritis (PsA).
In RA for example it activates synovial fibroblasts, which causes the overproduction of cathepsins and MMP.
The breakdown of collagen and proteoglycan follows, resulting in cartilage and bone destruction, as well as joint erosion.
These are: cytokines, chemokines, lipids, ROS / RNS, proteases and histamine.
So let’s break each one down now.
First we have pro-inflammatory cytokines.
These are small secreted proteins that amplify and coordinate inflammation.
The main ones are TNF-alpha, IL-1, IL-6, and IL-8.
TNF-alpha, is mainly secreted by (activated) macrophages and Th1 cells in response to bacterial lipopolysaccharide, exists as a transmembrane precursor cleaved by the enzyme TACE into a soluble trimer.
It plays key roles in the pathogenesis of autoimmune diseases such as rheumatoid arthritis (RA), IBD and psoriatic arthritis (PsA).
In RA for example it activates synovial fibroblasts, which causes the overproduction of cathepsins and MMP.
The breakdown of collagen and proteoglycan follows, resulting in cartilage and bone destruction, as well as joint erosion.



IL-1 exists in two forms: IL-1alpha and IL-1beta.
IL-1beta synthesis requires two signals: transcriptional upregulation via NF-kappaB, followed by inflammasome-mediated cleavage by caspase-1.
The inflammasome assembles in response to danger signals such as ATP, uric acid crystals, or bacterial toxins.
IL-6, secreted by macrophages, T cells, and fibroblasts, binds a membrane or soluble receptor and signals through gp130 and JAK-STAT3.
It is the primary driver of the hepatic acute-phase response, stimulates B-cell differentiation, and promotes Th17 cell development.
IL-8 produced by macrophages and endothelial cells, binds CXCR1 and CXCR2 receptors to attract and activate neutrophils (it dominates in bacterial infections, psoriasis and acute respiratory distress syndrome).
Other important cytokines include IL-12 and IL-23, which drive Th1 and Th17 responses, respectively and IL-17, which recruits neutrophils and induces defensins in psoriasis and spondyloarthritis.
IL-1beta synthesis requires two signals: transcriptional upregulation via NF-kappaB, followed by inflammasome-mediated cleavage by caspase-1.
The inflammasome assembles in response to danger signals such as ATP, uric acid crystals, or bacterial toxins.
IL-6, secreted by macrophages, T cells, and fibroblasts, binds a membrane or soluble receptor and signals through gp130 and JAK-STAT3.
It is the primary driver of the hepatic acute-phase response, stimulates B-cell differentiation, and promotes Th17 cell development.
IL-8 produced by macrophages and endothelial cells, binds CXCR1 and CXCR2 receptors to attract and activate neutrophils (it dominates in bacterial infections, psoriasis and acute respiratory distress syndrome).
Other important cytokines include IL-12 and IL-23, which drive Th1 and Th17 responses, respectively and IL-17, which recruits neutrophils and induces defensins in psoriasis and spondyloarthritis.
Then we have chemokines.
Chemokines are a subset of cytokines that function as chemotactic gradients to direct leukocyte migration during inflammation and are classified into four families (based on cysteine spacing): CXC, CC, C and CX3C.
The prototype neutrophil chemoattractant is IL-8, also known as CXCL8, produced by macrophages, endothelial cells, and neutrophils themselves in response to IL-1 or TNF-alpha.
It binds CXCR1 and CXCR2 receptors to trigger neutrophil degranulation, respiratory burst and firm adhesion via integrin activation.
MCP-1, or CCL2, recruits monocytes and memory T cells by binding CCR2 and is central to atherosclerotic plaque formation and chronic inflammation in obesity.
RANTES or CCL5, attracts eosinophils, basophils, and T cells via CCR1, CCR3, and CCR5 and plays a major role in allergic inflammation and viral infections.
Eotaxin, or CCL11, specifically draws eosinophils in asthma and parasitic infections.
Fractalkine, or CX3CL1, exists in membrane-bound and soluble forms; the membrane form promotes firm adhesion, while the soluble form acts as a chemoattractant for monocytes and NK cells.
Chemokine production is tightly regulated by NF-kappaB and occurs within hours of tissue injury, creating gradients that guide the ordered arrival of neutrophils first, followed by monocytes.
Chemokines are a subset of cytokines that function as chemotactic gradients to direct leukocyte migration during inflammation and are classified into four families (based on cysteine spacing): CXC, CC, C and CX3C.
The prototype neutrophil chemoattractant is IL-8, also known as CXCL8, produced by macrophages, endothelial cells, and neutrophils themselves in response to IL-1 or TNF-alpha.
It binds CXCR1 and CXCR2 receptors to trigger neutrophil degranulation, respiratory burst and firm adhesion via integrin activation.
MCP-1, or CCL2, recruits monocytes and memory T cells by binding CCR2 and is central to atherosclerotic plaque formation and chronic inflammation in obesity.
RANTES or CCL5, attracts eosinophils, basophils, and T cells via CCR1, CCR3, and CCR5 and plays a major role in allergic inflammation and viral infections.
Eotaxin, or CCL11, specifically draws eosinophils in asthma and parasitic infections.
Fractalkine, or CX3CL1, exists in membrane-bound and soluble forms; the membrane form promotes firm adhesion, while the soluble form acts as a chemoattractant for monocytes and NK cells.
Chemokine production is tightly regulated by NF-kappaB and occurs within hours of tissue injury, creating gradients that guide the ordered arrival of neutrophils first, followed by monocytes.

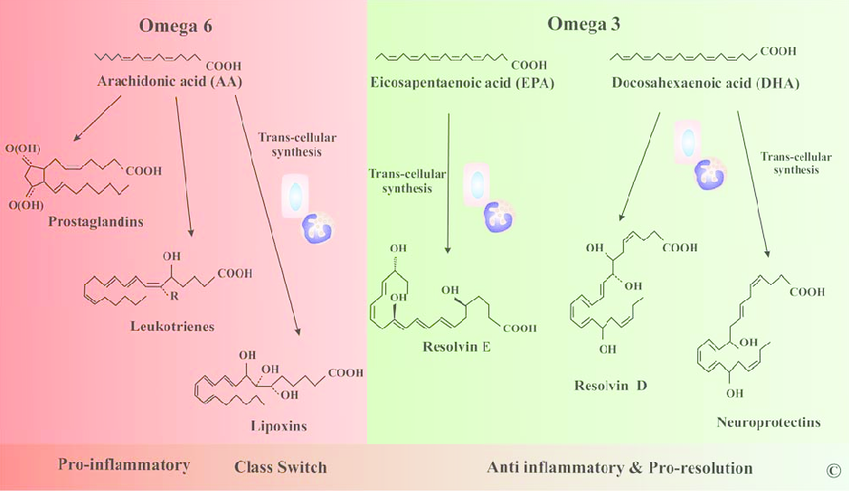
Moving on to lipid mediators.
Lipid mediators are rapidly generated from membrane phospholipids and act locally to modulate vascular tone, permeability, and leukocyte function.
All lipid mediators are derived from arachidonic acid (AA), released by phospholipase A2 (PLA₂) from cell membranes, have half-lives of seconds to minutes and are degraded by specific enzymes such as 15-hydroxyprostaglandin dehydrogenase.
The pathway begins with phospholipase A2 liberating arachidonic acid, which feeds into cyclooxygenase or lipoxygenase branches.
Prostaglandin E2, produced via inducible COX-2 and mPGES-1 in macrophages and fibroblasts, is the dominant lipid in acute inflammation.
It synergizes with histamine and bradykinin to cause vasodilation and pain sensitization through EP2 and EP4 receptors, induces fever via hypothalamic EP3 receptors and at later stages promotes resolution by switching macrophages to an M2 phenotype.
Prostacyclin, or PGI2, from endothelial cells potently vasodilates and inhibits platelet aggregation.
Leukotriene B4, generated via 5-lipoxygenase in neutrophils and macrophages, is a powerful neutrophil chemoattractant and activator that rivals IL-8 in potency.
Cysteinyl leukotrienes LTC4, LTD4, and LTE4, produced by mast cells and eosinophils, cause bronchoconstriction, mucus secretion and vascular leakage in asthma.
Lipid mediators are rapidly generated from membrane phospholipids and act locally to modulate vascular tone, permeability, and leukocyte function.
All lipid mediators are derived from arachidonic acid (AA), released by phospholipase A2 (PLA₂) from cell membranes, have half-lives of seconds to minutes and are degraded by specific enzymes such as 15-hydroxyprostaglandin dehydrogenase.
The pathway begins with phospholipase A2 liberating arachidonic acid, which feeds into cyclooxygenase or lipoxygenase branches.
Prostaglandin E2, produced via inducible COX-2 and mPGES-1 in macrophages and fibroblasts, is the dominant lipid in acute inflammation.
It synergizes with histamine and bradykinin to cause vasodilation and pain sensitization through EP2 and EP4 receptors, induces fever via hypothalamic EP3 receptors and at later stages promotes resolution by switching macrophages to an M2 phenotype.
Prostacyclin, or PGI2, from endothelial cells potently vasodilates and inhibits platelet aggregation.
Leukotriene B4, generated via 5-lipoxygenase in neutrophils and macrophages, is a powerful neutrophil chemoattractant and activator that rivals IL-8 in potency.
Cysteinyl leukotrienes LTC4, LTD4, and LTE4, produced by mast cells and eosinophils, cause bronchoconstriction, mucus secretion and vascular leakage in asthma.
When it comes to ROS / RNS, reactive oxygen species and reactive nitrogen species are generated by immune cells as microbicidal weapons but also amplify inflammation through tissue signaling.
Neutrophils and macrophages assemble the NADPH oxidase complex upon phagocytosis or Fc receptor engagement, producing superoxide that dismutates to hydrogen peroxide and then hypochlorous acid via myeloperoxidase.
These oxidants damage microbial membranes and DNA but also oxidize host lipids and proteins, creating danger signals that activate the NLRP3 inflammasome.
Peroxynitrite, formed by superoxide reacting with nitric oxide from inducible nitric oxide synthase in macrophages, nitrates tyrosine residues and disrupts endothelial barrier function.
ROS activate redox-sensitive transcription factors NF-kappaB and AP-1, upregulating cytokine and adhesion molecule expression, and induce mitochondrial damage that releases mtDNA to further stimulate inflammation.
In resolution, antioxidant systems such as superoxide dismutase, catalase, and glutathione peroxidase limit damage, while NRF2 activation drives cytoprotective gene expression.
Neutrophils and macrophages assemble the NADPH oxidase complex upon phagocytosis or Fc receptor engagement, producing superoxide that dismutates to hydrogen peroxide and then hypochlorous acid via myeloperoxidase.
These oxidants damage microbial membranes and DNA but also oxidize host lipids and proteins, creating danger signals that activate the NLRP3 inflammasome.
Peroxynitrite, formed by superoxide reacting with nitric oxide from inducible nitric oxide synthase in macrophages, nitrates tyrosine residues and disrupts endothelial barrier function.
ROS activate redox-sensitive transcription factors NF-kappaB and AP-1, upregulating cytokine and adhesion molecule expression, and induce mitochondrial damage that releases mtDNA to further stimulate inflammation.
In resolution, antioxidant systems such as superoxide dismutase, catalase, and glutathione peroxidase limit damage, while NRF2 activation drives cytoprotective gene expression.

Moving on to proteases.
Proteases released by inflammatory cells degrade extracellular matrix, activate pro-cytokines, and modulate receptor signaling.
Neutrophil elastase, stored in azurophilic granules and released during degranulation or NETosis, cleaves bacterial virulence factors but also host elastin, collagen, and complement components, contributing to tissue destruction in emphysema and abscess formation.
Matrix metalloproteinases, particularly MMP-9 from neutrophils and MMP-1 and MMP-3 from macrophages and synoviocytes, are transcriptionally induced by IL-1 and TNF-alpha and degrade collagen and gelatin, enabling leukocyte migration and driving joint erosion in arthritis.
Cathepsins from macrophage lysosomes process antigens and activate pro-IL-1beta.
The coagulation cascade protease thrombin not only forms fibrin but also signals through PAR-1 to induce endothelial permeability and chemokine production.
Proteases released by inflammatory cells degrade extracellular matrix, activate pro-cytokines, and modulate receptor signaling.
Neutrophil elastase, stored in azurophilic granules and released during degranulation or NETosis, cleaves bacterial virulence factors but also host elastin, collagen, and complement components, contributing to tissue destruction in emphysema and abscess formation.
Matrix metalloproteinases, particularly MMP-9 from neutrophils and MMP-1 and MMP-3 from macrophages and synoviocytes, are transcriptionally induced by IL-1 and TNF-alpha and degrade collagen and gelatin, enabling leukocyte migration and driving joint erosion in arthritis.
Cathepsins from macrophage lysosomes process antigens and activate pro-IL-1beta.
The coagulation cascade protease thrombin not only forms fibrin but also signals through PAR-1 to induce endothelial permeability and chemokine production.
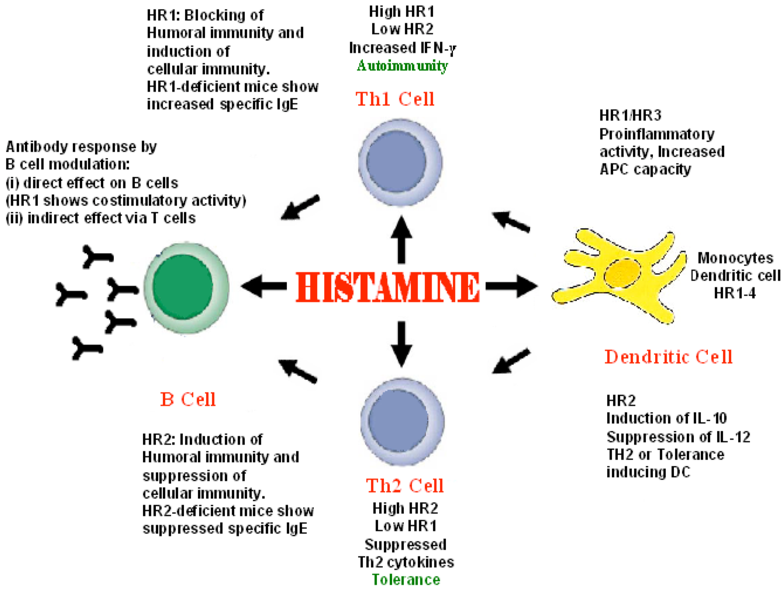
And finally, we have histamine.
Histamine is a preformed mediator stored in mast cell and basophil granules and released within seconds of IgE cross-linking, complement activation, or physical stimuli, which is rapidly metabolized by histamine N-methyltransferase and diamine oxidase (DAO).
It acts on H1 receptors to cause vasodilation, endothelial gap formation, and smooth muscle contraction, producing the immediate phase of increased permeability and wheal-and-flare response in urticaria.
H2 receptor activation increases gastric acid secretion and modulates immune responses. In early inflammation, histamine synergizes with lipid mediators to amplify edema and recruits eosinophils via H4 receptors.
Mast cell degranulation also releases tryptase, which activates PAR-2 to further increase permeability and cytokine production.
Histamine is a preformed mediator stored in mast cell and basophil granules and released within seconds of IgE cross-linking, complement activation, or physical stimuli, which is rapidly metabolized by histamine N-methyltransferase and diamine oxidase (DAO).
It acts on H1 receptors to cause vasodilation, endothelial gap formation, and smooth muscle contraction, producing the immediate phase of increased permeability and wheal-and-flare response in urticaria.
H2 receptor activation increases gastric acid secretion and modulates immune responses. In early inflammation, histamine synergizes with lipid mediators to amplify edema and recruits eosinophils via H4 receptors.
Mast cell degranulation also releases tryptase, which activates PAR-2 to further increase permeability and cytokine production.
So:
-Cytokines (TNF-α, IL-1, IL-6, IL-17 etc) -> macrophages and T cells.
-Chemokines (IL-8, MCP-1, RANTES etc) -> macrophages and endothelium.
-Lipid mediators (PGE₂, LTB4, PAF etc) -> macrophages and mast cells.
-ROS / RNS (superoxide, NO etc) -> neutrophils and macrophages.
-Proteases (elastase, MMP-9 etc) -> neutrophils and macrophages.
-Histamine -> mast cells and basophils.
-Cytokines (TNF-α, IL-1, IL-6, IL-17 etc) -> macrophages and T cells.
-Chemokines (IL-8, MCP-1, RANTES etc) -> macrophages and endothelium.
-Lipid mediators (PGE₂, LTB4, PAF etc) -> macrophages and mast cells.
-ROS / RNS (superoxide, NO etc) -> neutrophils and macrophages.
-Proteases (elastase, MMP-9 etc) -> neutrophils and macrophages.
-Histamine -> mast cells and basophils.
Now, here's a list of common lifestyle factors and conditions that exacerbate systemic inflammation:
-Being overweight increases everything (TNF-α, IL-6, IL-1β, MCP-1, IL-8 etc).
-Trans fats that (mainly) increase TNF-α and IL-6.
-A high O6, low O3 ratio in the diet that (mainly) increases PGE2 and IL-8.
-Anything that can lead to endotoxemia will (mainly) increase IL-6, TNF-α and IL-1β.
-Alcohol (more than 1 shot a day) will increase IL-6, TNF-α and IL-1β.
-T2D/high blood sugar can elevate IL-6 and CRP.
-Too much iron can increase IL-6 and CRP.
-Sleep deprivation increases IL-6, TNF-α, CRP.
-Sleep apnea increases IL-6, IL-8, IL-1β.
-Low muscle mass increases IL-6 and CRP.
-Sedentary behavior (>8 h/day) increases IL-6 and TNF-α.
-Chronic stress also increases pretty much everything.
-Being overweight increases everything (TNF-α, IL-6, IL-1β, MCP-1, IL-8 etc).
-Trans fats that (mainly) increase TNF-α and IL-6.
-A high O6, low O3 ratio in the diet that (mainly) increases PGE2 and IL-8.
-Anything that can lead to endotoxemia will (mainly) increase IL-6, TNF-α and IL-1β.
-Alcohol (more than 1 shot a day) will increase IL-6, TNF-α and IL-1β.
-T2D/high blood sugar can elevate IL-6 and CRP.
-Too much iron can increase IL-6 and CRP.
-Sleep deprivation increases IL-6, TNF-α, CRP.
-Sleep apnea increases IL-6, IL-8, IL-1β.
-Low muscle mass increases IL-6 and CRP.
-Sedentary behavior (>8 h/day) increases IL-6 and TNF-α.
-Chronic stress also increases pretty much everything.
-Mold mycotoxins increase IL-1β, IL-6 and TNF-α.
-A magnesium deficiency increases IL-6, CRP.
-A vitamin C deficiency increases IL-1β.
-A zinc deficiency increases IL-6 and IL-1β.
-A selenium deficiency increases TNF-α.
-A vitamin D deficiency increases IL-6, TNF-α and IL-17.
-Melatonin suppression pretty much also increases almost everything down the line.
-Heavy metals IL-6, IL-1β and TNF-α (but less than mold).
-Overtraining increases IL-6 and IL-1β.
-MCAS also increases pretty much everything.
-A magnesium deficiency increases IL-6, CRP.
-A vitamin C deficiency increases IL-1β.
-A zinc deficiency increases IL-6 and IL-1β.
-A selenium deficiency increases TNF-α.
-A vitamin D deficiency increases IL-6, TNF-α and IL-17.
-Melatonin suppression pretty much also increases almost everything down the line.
-Heavy metals IL-6, IL-1β and TNF-α (but less than mold).
-Overtraining increases IL-6 and IL-1β.
-MCAS also increases pretty much everything.
Now here are some tests you can take in order to "assess" your inflammation "status".
Tier 1: Non-negotiables
-hs-CRP (IL-6 driven) (the reason pomegranate/pomegranate extracts + PQQ are often recommended for cardiovascular health are partly because they lower CRP and IL-6 quite a lot and way more in people struggling with issues such as T2D).
-Vitamin and mineral status (an RBC element test can also be added and vitamin D should be included (even 25-hydroxy vitamin D)).
-CBC (neutrophils, lymphocytes, monocytes).
-Erythrocyte sedimentation rate.
-Oxidized LDL.
-Procalcitonin.
Tier 1: Non-negotiables
-hs-CRP (IL-6 driven) (the reason pomegranate/pomegranate extracts + PQQ are often recommended for cardiovascular health are partly because they lower CRP and IL-6 quite a lot and way more in people struggling with issues such as T2D).
-Vitamin and mineral status (an RBC element test can also be added and vitamin D should be included (even 25-hydroxy vitamin D)).
-CBC (neutrophils, lymphocytes, monocytes).
-Erythrocyte sedimentation rate.
-Oxidized LDL.
-Procalcitonin.
Tier 2: Chronic health issues
-Cytokine panel.
-Th1/Th2/Th17 balance.
-24h urine n-methylhistamine.
-Anticcp / rf.
-24h urine prostaglandin D2.
-Fecal zonulin.
-24h urine leukotriene E4.
Then based on the results, you can further dial in your approach after you've covered the common lifestyle factors and conditions that exacerbate systemic inflammation.
-Cytokine panel.
-Th1/Th2/Th17 balance.
-24h urine n-methylhistamine.
-Anticcp / rf.
-24h urine prostaglandin D2.
-Fecal zonulin.
-24h urine leukotriene E4.
Then based on the results, you can further dial in your approach after you've covered the common lifestyle factors and conditions that exacerbate systemic inflammation.
OTC tools that you can further look into in regards to inflammation include:
-BPC-157
-Luteolin
-Quercetin
-Whole food vitamin E
-B vitamins
-PQQ
-Aspirin + K1/K2
-CBD
-Glutathione (injectable)
-Ubiquinol
-Taurine
-Curcumin
-Apolactoferrin
-Blackseed oil
-BPC-157
-Luteolin
-Quercetin
-Whole food vitamin E
-B vitamins
-PQQ
-Aspirin + K1/K2
-CBD
-Glutathione (injectable)
-Ubiquinol
-Taurine
-Curcumin
-Apolactoferrin
-Blackseed oil




And of course, don't forget about grounding.

That's all.
For more on the topic of hair loss, go here: fitandball.gumroad.com/l/dfgdfgdfgfg/…
For more on the topic of hair loss, go here: fitandball.gumroad.com/l/dfgdfgdfgfg/…
And if you learned something from this thread, make sure to leave a like/RT.
https://x.com/Helios_Movement/status/2066890494743884082
• • •
Missing some Tweet in this thread? You can try to
force a refresh