In 1956, Benjamin Castleman et al described 13 cases of mediastinal lymph node hyperplasia that resembled thymomas grossly and microscopically 


Castleman argued against their thymic or neoplastic origin.
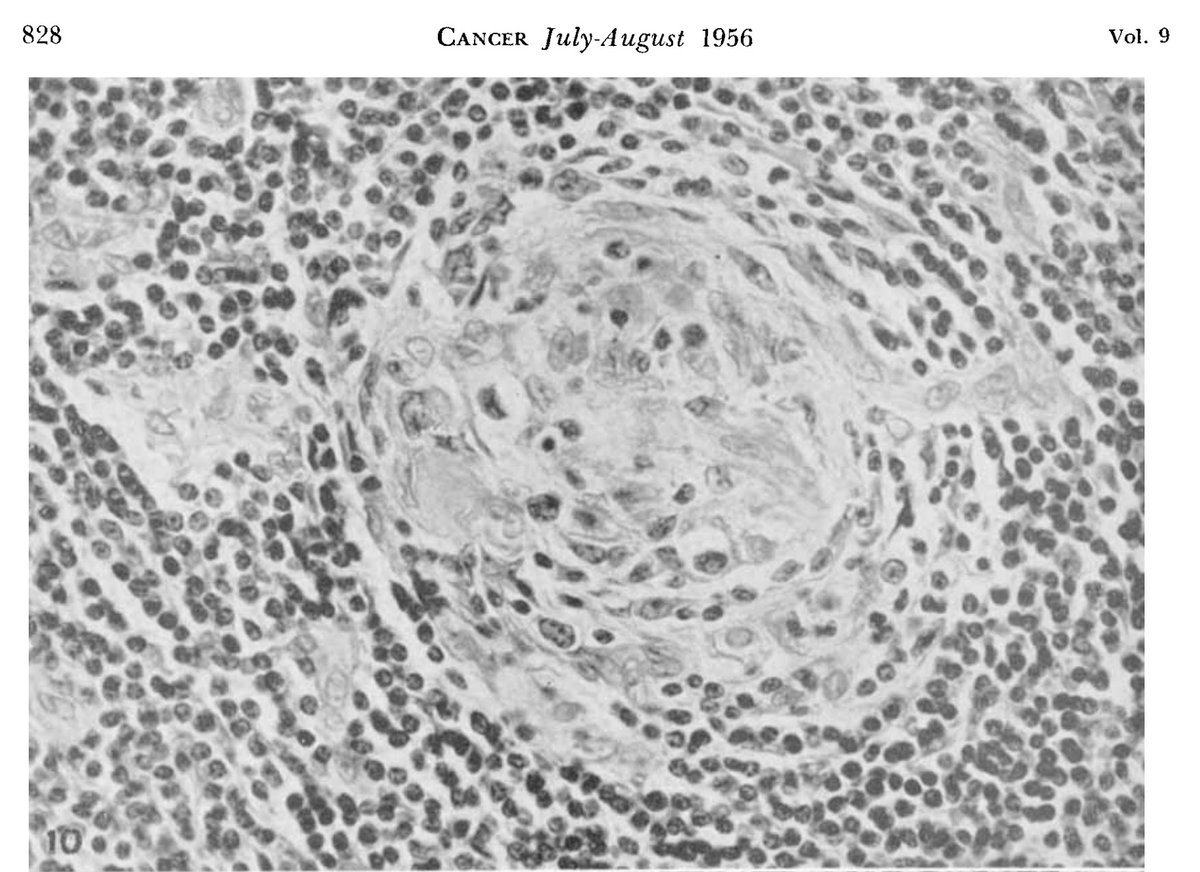
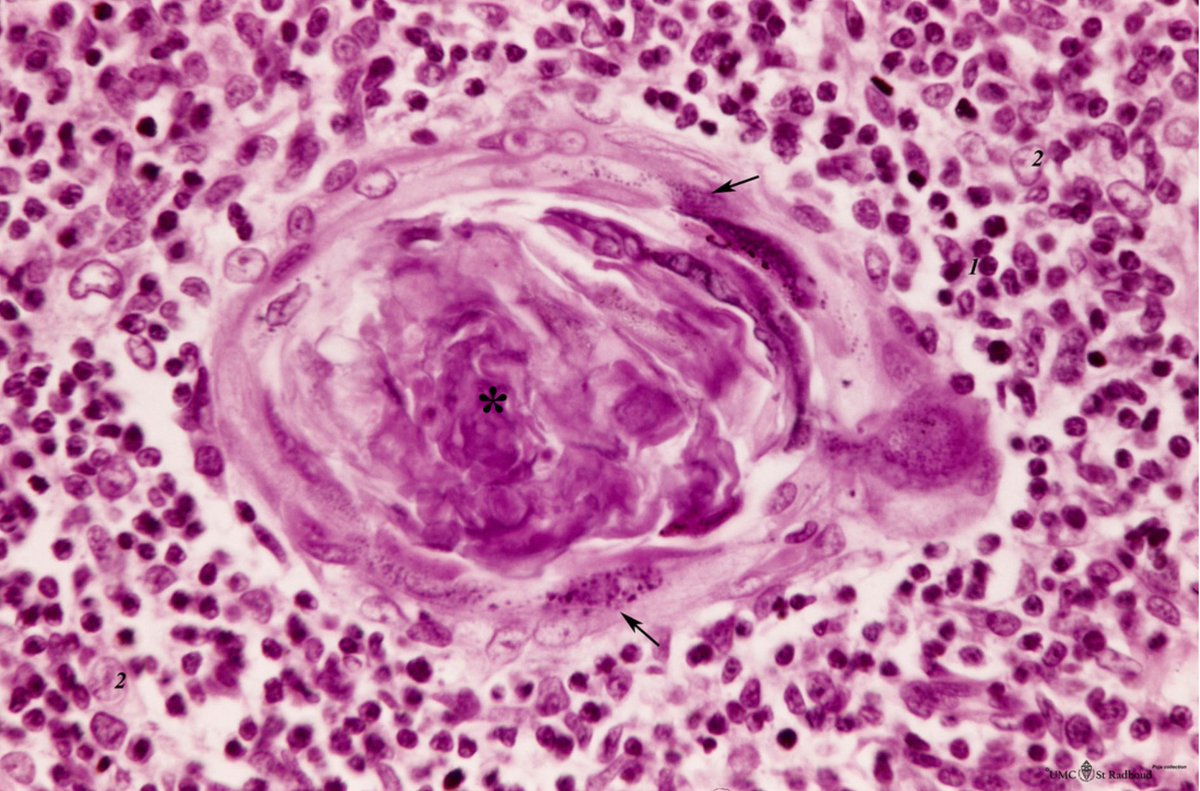
The nodes were highly vascular with marked capillary proliferation with hyaline thickening of the vessel walls penetrating the follicles (left), resembling Hassall corpuscles (right).
The nodes were highly vascular with marked capillary proliferation with hyaline thickening of the vessel walls penetrating the follicles (left), resembling Hassall corpuscles (right).
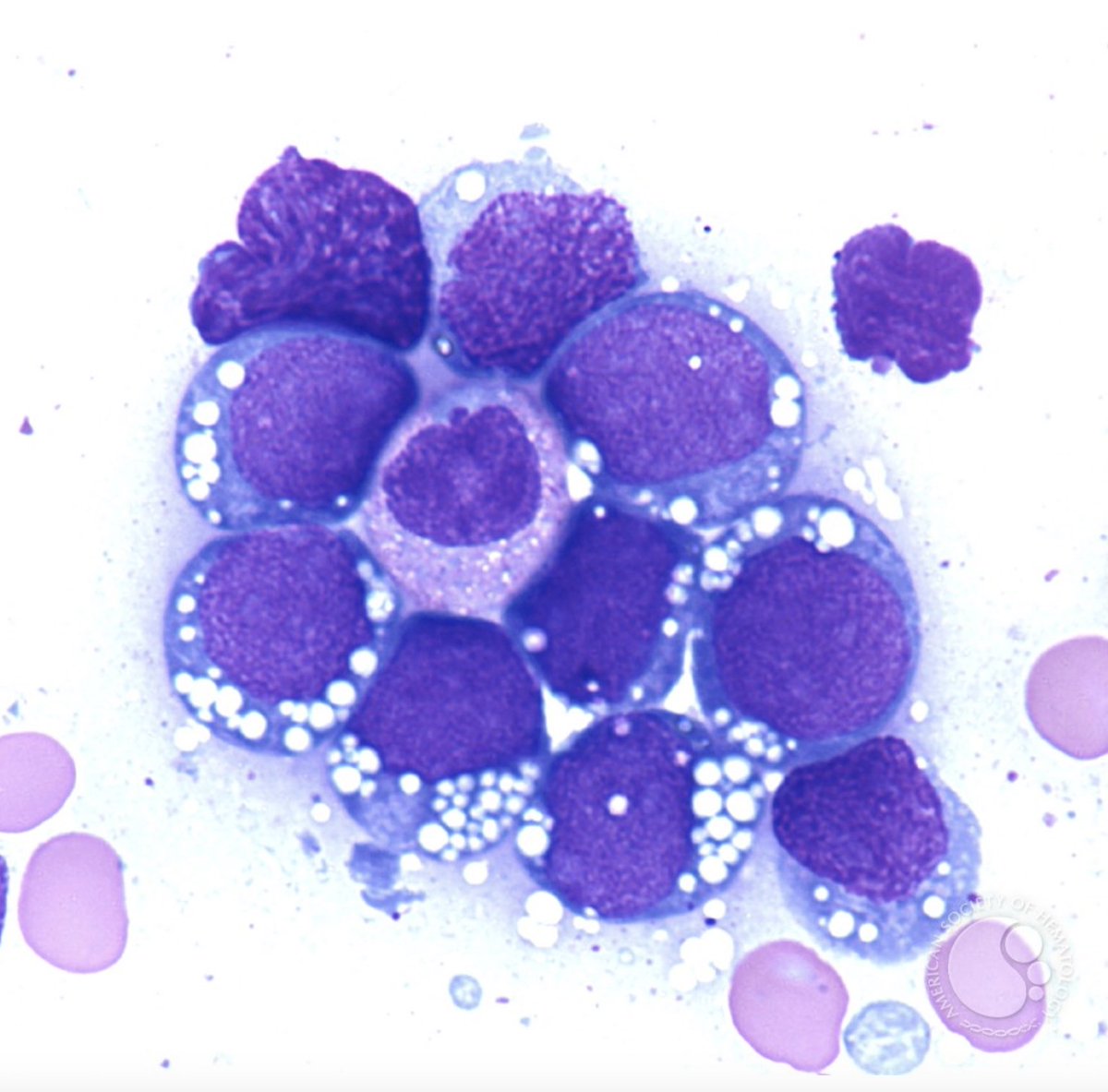
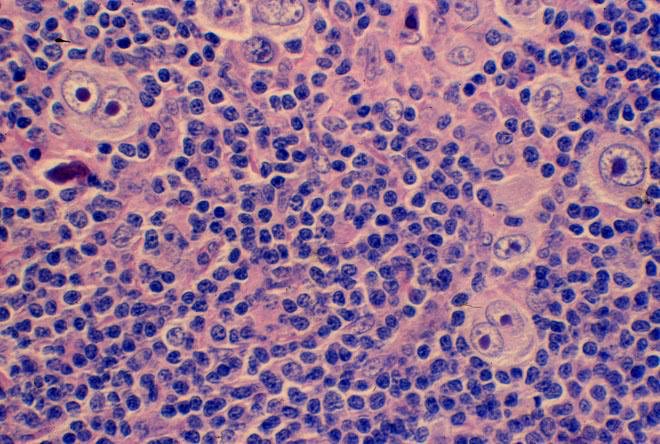
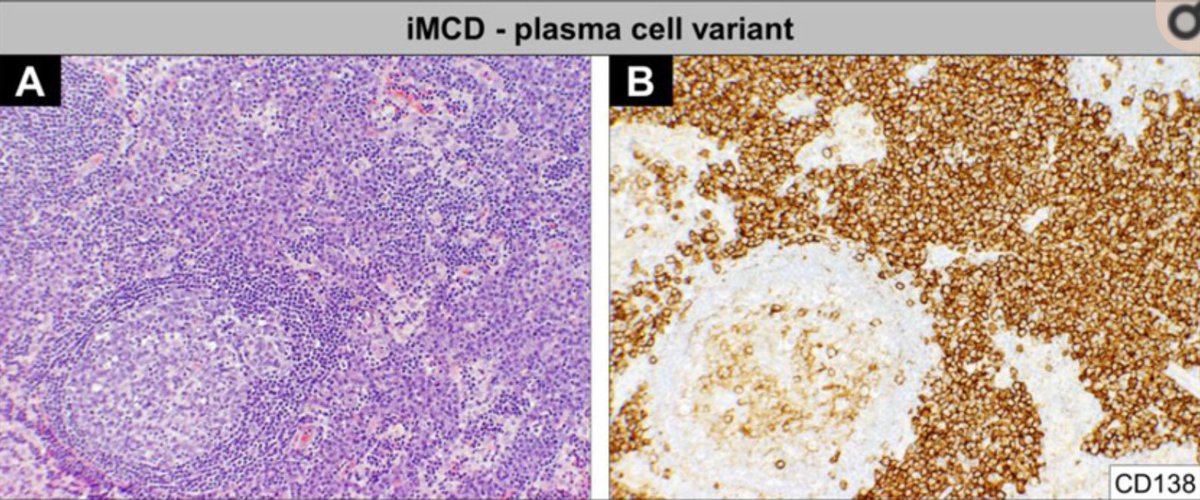
In about half, the lymph node hyperplasia demonstrated follicular hyperplasia and interfollicular sheets of mature plasma cells.
Most patients had no symptoms and were essentially cured with surgical resection.
Most patients had no symptoms and were essentially cured with surgical resection.
Over the next 10 years, dozens of cases were reported, many of which were located in the mediastinum. They almost exclusively had a “hyaline vascular” appearance.
This entity was also referred to as “angiofollicular lymph node hyperplasia” and “giant lymph node hyperplasia”
This entity was also referred to as “angiofollicular lymph node hyperplasia” and “giant lymph node hyperplasia”


Some patients, however, did have systemic symptoms (fever, night sweats), anemia, hyper-gammaglobulinemia. Those cases were more likely to have a “plasma cell” appearance histologically.
This distinction was further established by Castleman et al in 1972 after reviewing 81 case
This distinction was further established by Castleman et al in 1972 after reviewing 81 case

By the 1970s cases of multicentric “angiofollicular lymph node hyperplasia” were increasingly recognized.
It became clear that there were 2 distinct clinical subtypes of this entity - termed “unicentric”and “multicentric” Castleman’s disease.
It became clear that there were 2 distinct clinical subtypes of this entity - termed “unicentric”and “multicentric” Castleman’s disease.
In 1983, multicentric Castleman’s disease (MCD) was described in association with Kaposi’s sarcoma in a case report and “speculation”; something that you will rarely see in journals nowadays because it lacks a “p value”
The following year, 2 more cases were described.
The following year, 2 more cases were described.


In 1985, Frizzera et al described a series of 15 patients with MCD, and highlighted the common presence of constitutional symptoms, organomegaly, elevated ESR, cytopenias, and hypergammaglobulinemia.

Treatment of MCD was poorly defined. In Frizzera’s report, treatment most commonly consisted of corticosteroids, but a number of patients received chemotherapy (chlorambucil, vincristine, CVP). 1 patient had rapidly fatal disease, but the majority had persistent or recurrent dx.

In 1994, Chang et al, identified herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Termed HHV-8.
In 1995, these DNA sequences were detected in patients with multicentric Castleman’s disease and HIV. They were also found in some without HIV.
In 1995, these DNA sequences were detected in patients with multicentric Castleman’s disease and HIV. They were also found in some without HIV.


This association between HHV-8 and MCD became an important distinction in classifying the disease.
Patients with HHV-8 associated MCD seemed to have a higher incidence of non-Hodgkin lymphoma, estimated at 15-fold higher than HIV+ population.
Patients with HHV-8 associated MCD seemed to have a higher incidence of non-Hodgkin lymphoma, estimated at 15-fold higher than HIV+ population.
Castleman’s disease was thought to be a reactive lymphoproliferative disorder when it was first described.
This was confirmed by analysis of clonality in Castleman’s disease in 1995.
This was confirmed by analysis of clonality in Castleman’s disease in 1995.

For more on clonality, see @DavidSteensma’s excellent #HematologyTweetstory
HHV-8 negative MCD was termed “idiopathic” (i.e. without clear association).
What became clear in 1989 is that IL-6 played an important role driving an inflammatory state that, at its worse, represented a cytokine storm with multiorgan failure and death.
What became clear in 1989 is that IL-6 played an important role driving an inflammatory state that, at its worse, represented a cytokine storm with multiorgan failure and death.

Attempts to further categorize idiopathic MCD came from Japan when Kojima et al described 28 cases in 2008 that were divided into “plasmacytic lymphadenopathy” and “non-plasmacytic” that resembled the “hyaline vascular” variant seen with unicentric CD.

Idiopathic MCD is heterogeneous. Some patients have an associated POEMS syndrome with polyneuropathy and a monoclonal gammopathy, while others developed the so called TAFRO syndrome first described by Takai et al in 2010 in Japanese patients.

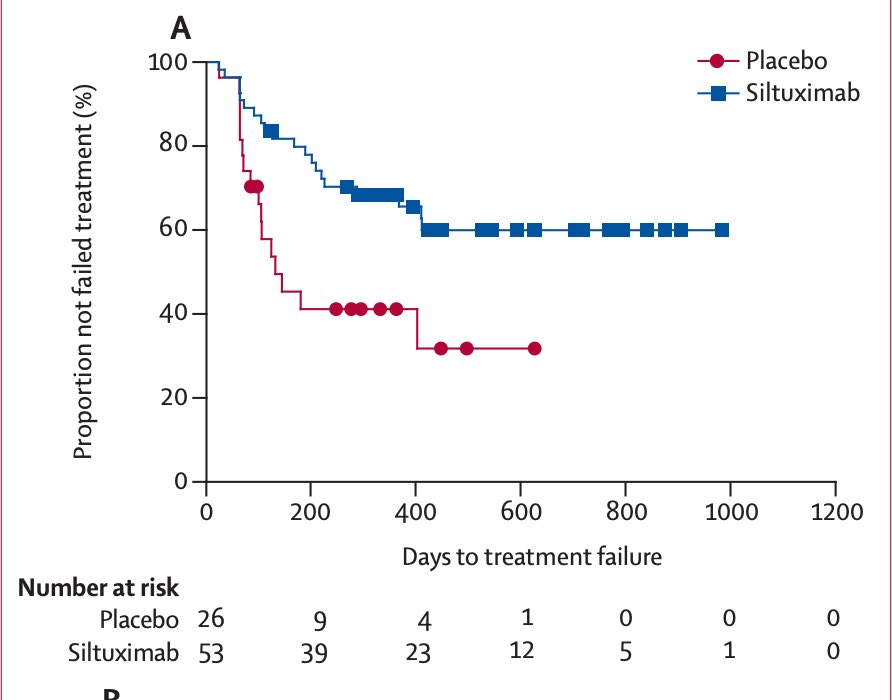
Around that time in 2010, the first (and only) randomized, placebo-controlled trial in idiopathic MCD started enrolling patients in 38 countries.
By 2014, siltuximab, a chimeric monoclonal antibody against IL-6, was approved by the @US_FDA
By 2014, siltuximab, a chimeric monoclonal antibody against IL-6, was approved by the @US_FDA

The newly formed Castleman Disease Collaborative Network (CDCN) led by @DavidFajgenbaum with a goal to accelerate CD and other rare disease research published consensus diagnostic criteria for idiopathic MCD in 2017.

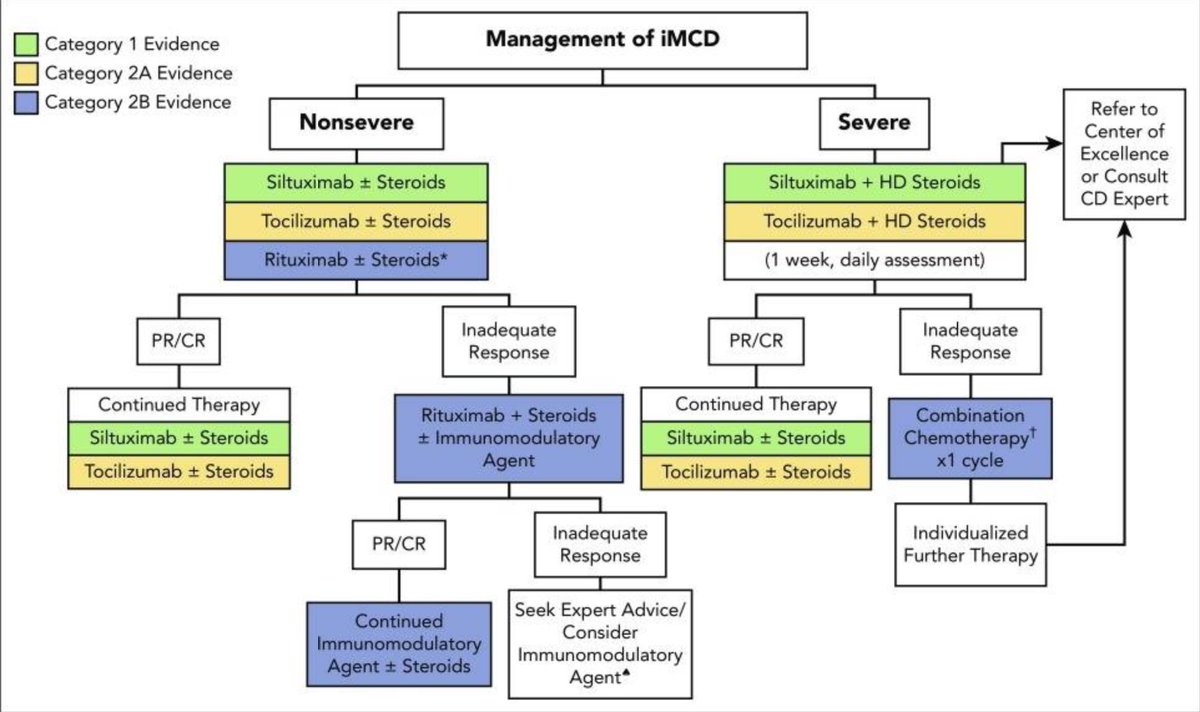
By 2018, the first treatment guidelines for idiopathic MCD were established incorporating experience from 60 years of published literature.
Castleman’s disease is one of many orphan diseases, with an estimated 5,000 cases diagnosed per year, and 30,000 patients living with some form of CD in the U.S. Like many rare diseases, CD is often misdiagnosed causing delay in initiating therapy in a timely manner.
@DavidFajgenbaum details his journey with idiopathic MCD in his book “Chasing My Cure”, which serves as a reminder that greater research focus, funding, and awareness are needed to make advances in the care of patients with CD and rare diseases in general.