
biorxiv.org/content/10.110…
In this Mmmanuscript we use our new tool M to break a couple of things, as is customary in #cryoEM. Most importantly, we show that 3.7 Å resolution can be achieved for a protein directly inside a cell. Software release next week. Now, thread:
In this Mmmanuscript we use our new tool M to break a couple of things, as is customary in #cryoEM. Most importantly, we show that 3.7 Å resolution can be achieved for a protein directly inside a cell. Software release next week. Now, thread:
M performs multi-particle refinement. One of single-particle analysis' central assumptions is that the particles are, in fact, single. This is physically never the case, and there is benefit in treating all items in a micrograph/tomogram as a connected system. 

This way deformation occurring during data acquisition can be modeled to improve registration of every particle's motion. Others* have done similar things over the years. M offers a unified framework for these concepts and aligns everything in one go.
M co-exists with Warp (duh!) and RELION, taking their alignments, improving them, and producing final high-res maps. Alternatively, you can take the improved alignments and go back to classify things more cleanly or attempt to find and solve other proteins in case of in situ.

Since M's deformation model and refinement capabilities are the same for frame and tilt series data, we were wondering: Is there any resolution penalty at all from using tilt series data? Sure, they're necessary for in situ stuff, but do we lose anything?
Turns out, we probably don't! @c_dienemann acquired equal amounts of apoferritin frame and tilt series data from neighboring positions on the same grid, and both data sets went to the same (record-setting for tilt series 😎) 2.3 Å resolution.

But maybe M's frame series processing just sucks, and that's why it matches the tilt series result? Luckily, #CAFR is happy to certify that M can produce competitive results for frame series data, too – achieving a 0.2 Å improvement to reach 1.34 Å on EMPIAR-10248.

Those who saw me at the CCP-EM symposium may remember a lower-resolution result shown there. Thanks to @biochem_fan's suggestion, Ewald sphere correction is a last-minute addition, available for both frame and tilt series refinement.
In situ imaging is my personal reason for staying in structural biology and developing #cryoEM methods. The prospect of being able to do 'visual systems biology' one day is very appealing, but we are still very, very far away from that.

I was delighted when Julia Mahamid and her student @Liang_Xue_ took a big leap of faith and decided to take the time to apply the Warp–RELION–M pipeline to their in situ tilt series. Liang had acquired data of whole M. pneumoniae cells and wanted to look at the 70S ribos inside.
They got ca. 11 Å using their old pipeline, so I figured 5–6 Å would be great. I couldn't quite believe my eyes when the resolution just kept climbing to 3.7 Å, almost reaching the Nyquist limit of the data.

The data were actually a control experiment for another study (biorxiv.org/content/10.110…), and the bacteria were drugged with chloramphenicol to stall the ribosomes. So we fitted the next best 70S+Cm PDB from E. coli, and there it was: Cm (in green), imaged inside a pathogen.
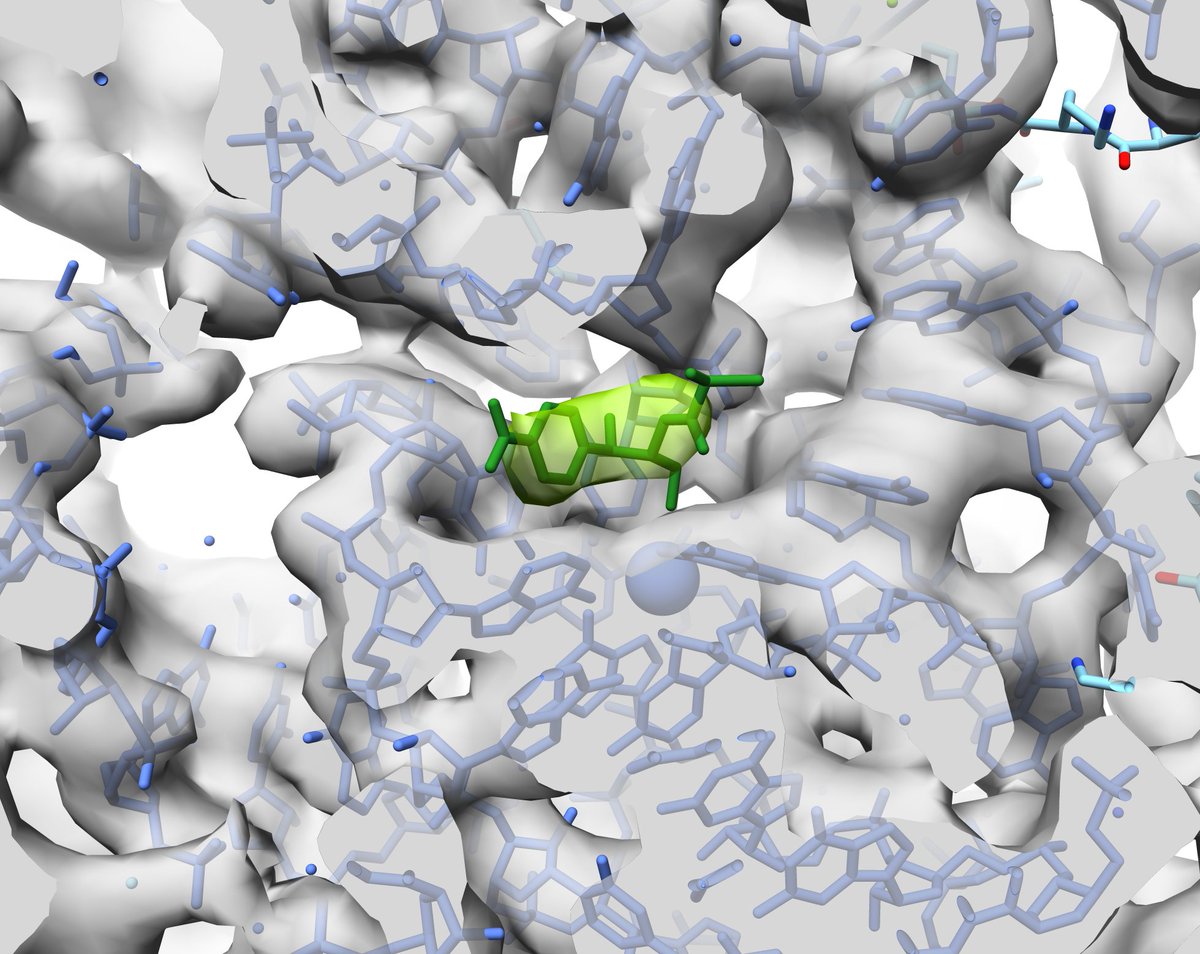
So that's exciting! But where do we go from here? It's tempting to think that 3.7 Å ribos mean similar in situ resolution for other proteins in the near future, as was historically the case for in vitro samples.
Unfortunately, in situ is a very different beast. Ribosomes are the perfect test object because they're large, easy to find by eye in tomograms, and very abundant. Pretty much everything else isn't, and that's the biggest bottleneck.
It will take a LOT more work by many more people to solve these issues or find ways to work around them. But at least image alignments don't seem like a big issue anymore, and that's what we celebrate today!
Image © belongs to the god of in situ visualization, @dsgoodsell
*This includes RELION's polishing, Alberto Bartesaghi's 2012 FREALIGN mod, and emClarity.

