
Excited to announce a new preprint! We did a study comparing two different @nanopore library prep approaches (ligation and rapid) for bacterial genomes with small plasmids:
biorxiv.org/content/10.110…
(1/11)
biorxiv.org/content/10.110…
(1/11)
I really like this paper because it has a clear conclusion simple enough to fit in a tweet: rapid preps are better than ligation preps at recovering small plasmids.
(2/11)
(2/11)
Figure 1 gives a simplified illustration of why we think this is the case: due to their size, small circular plasmids can avoid fragmentation during DNA extraction, leaving no ends for adapter ligation. Rapid preps, in contrast, don't depend on DNA ends.
(3/11)
(3/11)

Figure 2 shows our main results: using Illumina reads to approximate true plasmid abundance, the smaller a plasmid is, the more underrepresented it will likely be in a ligation read set. But not in a rapid read set!
(4/11)
(4/11)
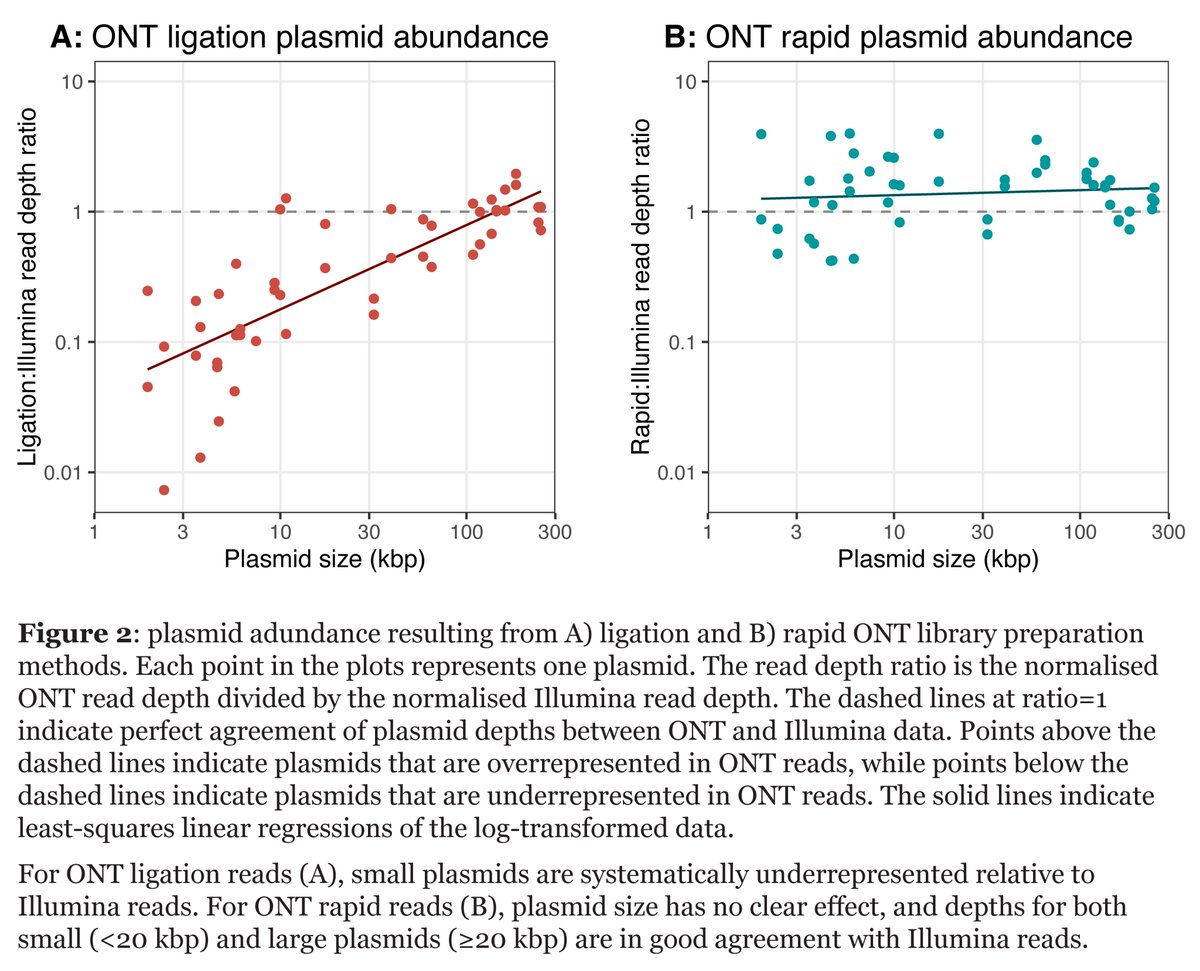
I like the plots in Figure 2 because the effect is obvious - I didn't even think it was necessary to do statistics (xkcd.com/2400). But I reluctantly did do linear regressions 😄
(5/11)
(5/11)
We also found another interesting difference between ligation and rapid: there were fewer chimeric reads in the rapid sets. This makes sense because rapid preps don't involve ligase, so there is less chance of combining two DNA fragments together.
(6/11)
(6/11)
So the advantages to using ligation preps are: better yield, more versatility and greater multiplexing of samples. For these reasons, it's what the @DrKatHolt lab mostly uses for Nanopore sequencing of bacterial isolates.
(7/11)
(7/11)
The advantages to using rapid preps are: simpler/faster procedure, potential for longer reads (if you're careful with DNA extraction), better representation of small plasmids and fewer chimeras.
(8/11)
(8/11)
The main takeaway of the paper is this: if you're doing Nanopore-only sequencing of a bacterial isolate and small plasmids matter to you, we recommend rapid preps! If you use a ligation prep, you might miss the small plasmids.
(9/11)
(9/11)
If you're doing hybrid (Nanopore+Illumina) sequencing, then ligation should be fine because the small plasmids will be captured by the Illumina reads. But expect the small plasmids to be underrepresented in your Nanopore reads.
(10/11)
(10/11)
Many thanks to the co-authors (@JuddLmj, @KelWyres and @DrKatHolt) and all the other members of the Holt Lab. And more generally, thanks to all the researchers out there who help make @nanopore bacterial genomics so good!
(11/11)
(11/11)
• • •
Missing some Tweet in this thread? You can try to
force a refresh




