Want to *see* how a tumour has evolved and grown? And also how different clones acquired characteristic transcriptional and histopathological features?
Hold on, that's magic.
No, it's our new preprint by @LucyYat47076319 and @MatsNilssonLab 1/9
biorxiv.org/content/10.110…
Hold on, that's magic.
No, it's our new preprint by @LucyYat47076319 and @MatsNilssonLab 1/9
biorxiv.org/content/10.110…

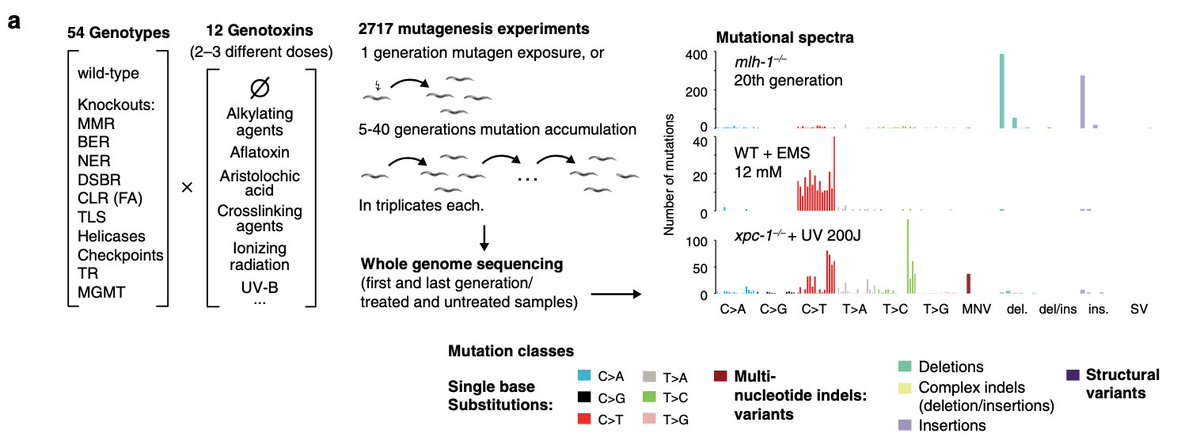
Jessica Svedlund developed a base-specific extension of the in situ sequencing protocol (BaSISS) to detect somatic mutations on a microscopy slide with fluorescently tagged padlock probes. 2/9

These signals are denoised and assembled into microscopic maps of subclonal growth using @LomakinAI's rigorous machine learning model. 3/9
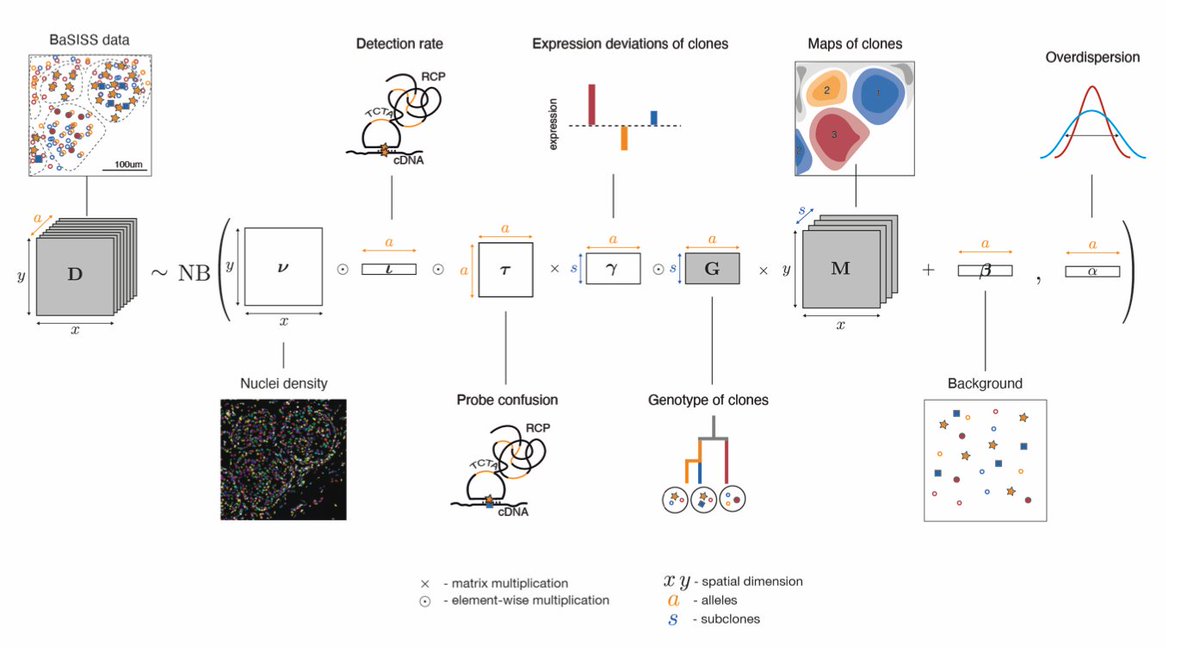
The results are eye watering (for me at least) and show the record of a raging competition of breast cancer clones. The observed clonal growth tightly followed physiological tissue structures. 4/9
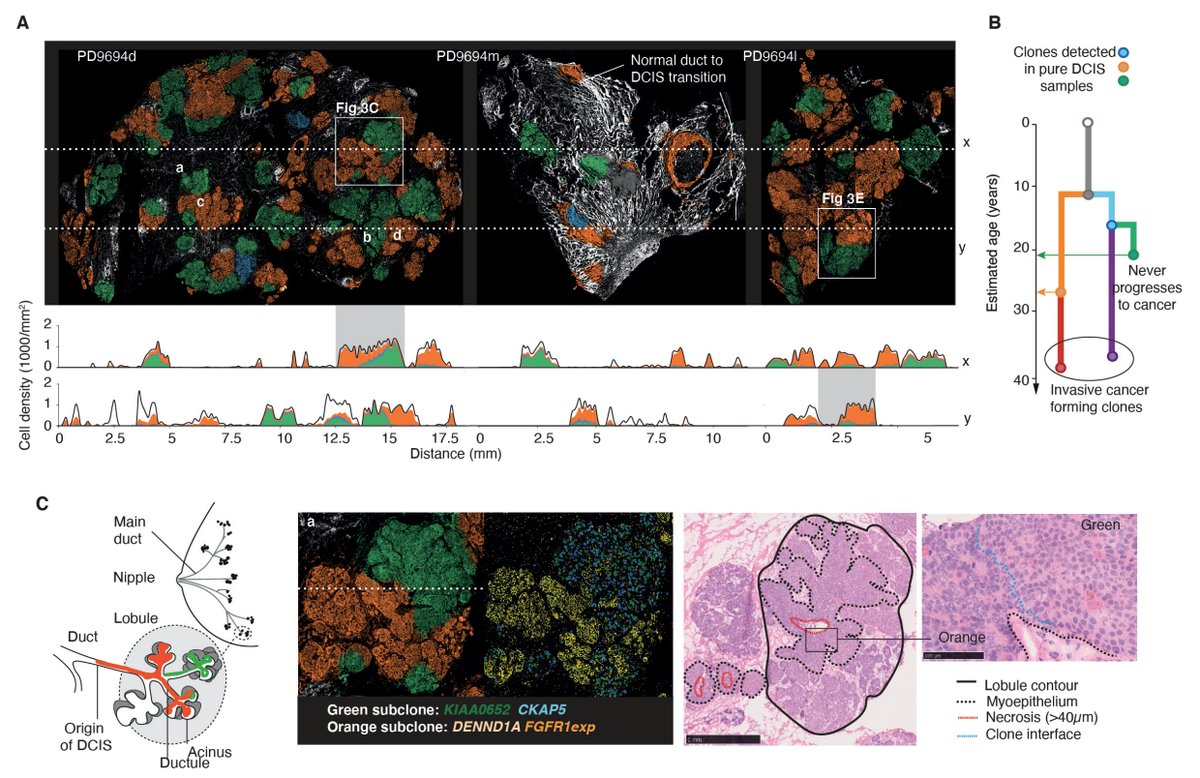
What’s more, layering in ISS probes targeting gene transcripts reveals each clone’s gene expression program and what type of micro environment it surrounds itself with. 5/9
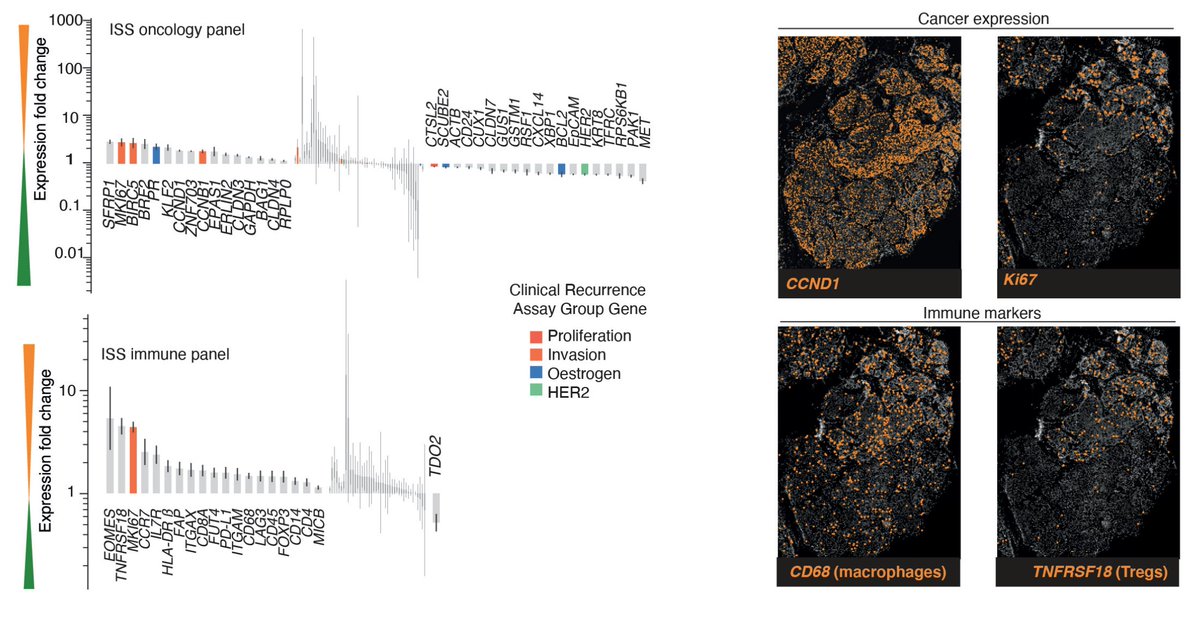
Clones can also have distinct histopathological appearance, further corroborating their phenotypic divergence. 6/9

Mapping these phenotypic patterns onto the underlying phylogeny brings an order into the observed heterogeneity and reveals directional changes as a tumour progresses. 7/9

These snippets highlight the huge potential of spatial genomics and transcriptomics with BaSISS to study cancer evolution. This will enable us to measure, understand - and hopefully one day prevent - the key steps of malignant progression. 8/9
It's the result of a great research collaboration combining new technology, clinical oncology and data science. Huge thanks to Carina, Milana, @artem_shmatko, Jun, @GenomeDoctor, @stefan_seq, @vitaliikl, Vasyl, Tong, @bayraktar_lab, @luiza_moore, Sarah, Andrea and Peter. 9/9
• • •
Missing some Tweet in this thread? You can try to
force a refresh