I am thrilled to announce that our work on In silico saturation mutagenesis of cancer genes is finally out @Nature. We propose a method inspired in evo biology to identify driver mutations in cancer genes. Here’s #tweetorial to sketch our results rdcu.be/cqsM1
Although we have a good knowledge of the genes that cause cancer upon mutations, most mutations in such genes are of uncertain significance. Classifying these variants in cancer genes is key to understand the mechanisms of tumorigenesis and advance precision medicine of cancer. 

One approach to bridge this gap is saturation mutagenesis (aka saturation genome editing), an amazing endeavour, at the moment done for a few cancer genes (e.g. TP53, PIK3CA, PTEN, BRCA1, Ras domain) in specific experimental systems.
Experimental saturation mutagenesis is costly and laborious, which has limited it to a handful of cancer genes and experimental systems, and the readout of which does not directly yield the oncogenic potential of mutations in human tissues.
We propose that mutations observed in thousands of human tumors, which constitute natural experiments testing their oncogenic potential in human tissues, can be exploited to learn their features and classify variants of uncertain significance (VUS) in cancer genes.
We set off by asking whether this question might be approachable by learning from the large catalogue of mutations in cancer genes across 28,000 tumors comprising 66 cancer types compiled and curated by intogen.org

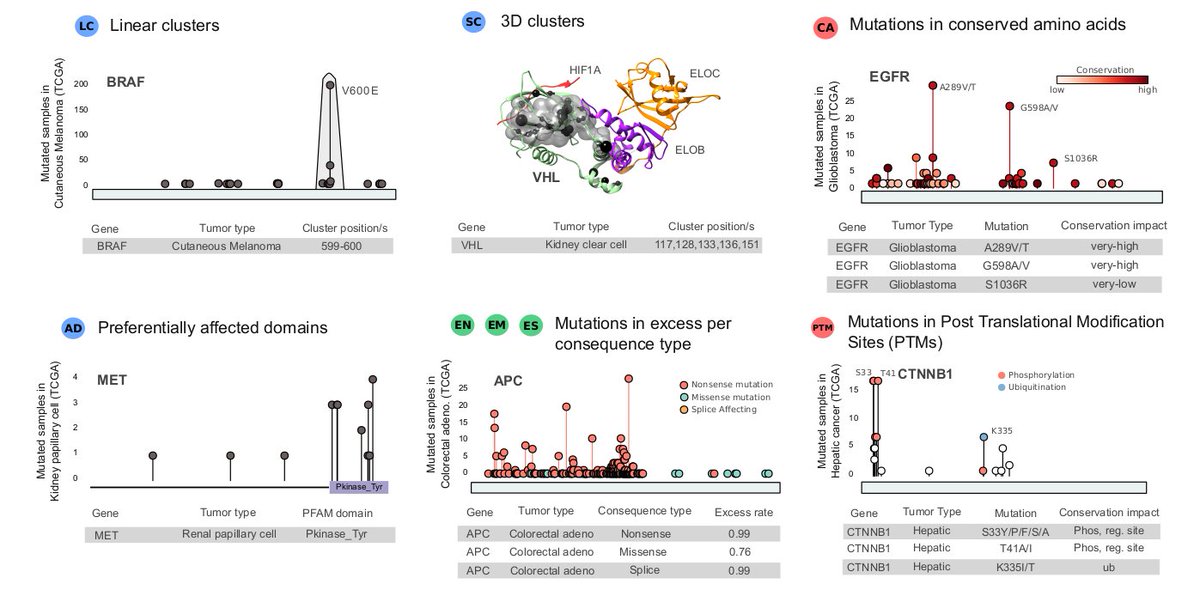
We assembled a training dataset with mutational features computed by IntOGen-pipeline for each cancer gene/tumor type, including clusters/hotspots, domains enriched by mutations, biases towards mutation consequences, plus others such as post-translational modif. and conservation
We devised a classification method based on a consensus of base models (gradient boosting) to distinguish between observed mutations matching a high dN/dS (i.e. highly enriched in driver mutations) and background neutral mutations, in a gene and tumor type specific manner.
To have a better sense of the bounds of learnability from our mutation data, we asked whether for each gene and cancer type we had observed a representative set of mutations to learn from. We thus defined a discovery index based on the unique mutation count in subsamples.

185 gene/tumor type pairs had enough mutations and good representativeness, and the models reached a good cross-validation performance, implying high reliability. This proof of concept suggests that richer mutational datasets might lead to good models for other gene/tumor types.


The models are designed to be interpretable, thus avoiding a black-box prediction device. We can derive additive explanation models which split the forecast in terms of relative contributions of the features (SHAP), thereby guiding the interpretation of the underlying mechanisms.


We validated boostDM models by holding out experimentally validated rare mutations then looking at the model predictions. BoostDM showed a high overall performance, with weighted F-scores above 0.9.
BoostDM models also outperformed available experimental saturation mutagenesis assays (5 genes) and state-of-the-art bioinformatic scores in mutations not included in model training.

We use specific and highly-reliable models to classify each possible mutation in a cancer gene in a tissue, producing what we call the blueprint of driver mutations.

BoostDM models can produce different blueprints of driver mutations in a gene in different tumor types (such as EGFR in lung adenocarcinoma and glioblastoma), reflecting divergence in tumorigenic mechanisms.

The blueprints uncover many potential driver mutations that have never been observed in tumors. So we asked whether the underlying mutational probability may contribute to defining the set of observed driver mutations.
Using the distribution of potential driver mutations in cancer genes we explore the interplay between the background mutagenesis and selection across tissues. We found a significant + bias across most cancer genes, overall greater in tumor-suppressor genes than in oncogenes.

To conclude, we show that the in silico saturation mutagenesis is a useful proof-of-concept to look closely into the mechanisms of tumorigenesis in a systematic way with a good outlook for improvement as new sequenced tumours are available and new features are considered.
The outputs of this work are relevant to the precision medicine of cancer. The Cancer Genome Interpreter (cancergenomeinterpreter.org) now feeds on the outputs of the in silico saturation mutagenesis for casting interpretations of single nucleotide variants.


The results of the in silico saturation mutagenesis for 185 high-quality models + 63 more general models are available for browsing here: intogen.org/boostdm
Reproducibility is essential for science to advance. We invested a decent amount of energy to make our analyses and methods as transparent and reproducible as possible.
A version of our pipeline is available here: bitbucket.org/bbglab/boostdm.
The code to re-generate all the figures of the paper is available here: github.com/bbglab/boostdm…
All the source data necessary for running the repos can be downloaded from zenodo: zenodo.org/record/4813082
The code to re-generate all the figures of the paper is available here: github.com/bbglab/boostdm…
All the source data necessary for running the repos can be downloaded from zenodo: zenodo.org/record/4813082
This is a team effort: full credit to co-authors @fran_mj88, @oriol_pich, Abel Gonzalez-Perez and @nlbigas for their huge investment in this project. What a privilege to be part of this team!

Also thanks to Iker Reyes-Salazar, @LorisMularoni, @miggrau, @dmartmillan, @ETapanari and @clarnedo for their technical contributions. And to the @bbglab at large for being such a stimulating and supportive community.
Last but not least, this kind of analysis would have never been possible without the generosity of donors, clinicians and institutional efforts to collect and sequence tumor samples. Shout out to @cbioportal, @HartwigMedical, @icgc_dcc, #PCAWG, @StJude and #TCGA.
• • •
Missing some Tweet in this thread? You can try to
force a refresh