
Absolutely thrilled to share our VASA-seq pre-print on #biorxiv today: Droplet-based single-cell total RNA-seq reveals differential non-coding expression and splicing patterns during mouse development 🧵
biorxiv.org/content/10.110…
biorxiv.org/content/10.110…
VASA-seq captures most parts of the transcriptome (total RNA) of a #singlecell rather than just a portion. It was expertly developed by Fredrik Salmen, a postdoc in @AlexandervanOu1's lab at the @_Hubrecht and partner in crime for this project 🕵️ (1/n). 
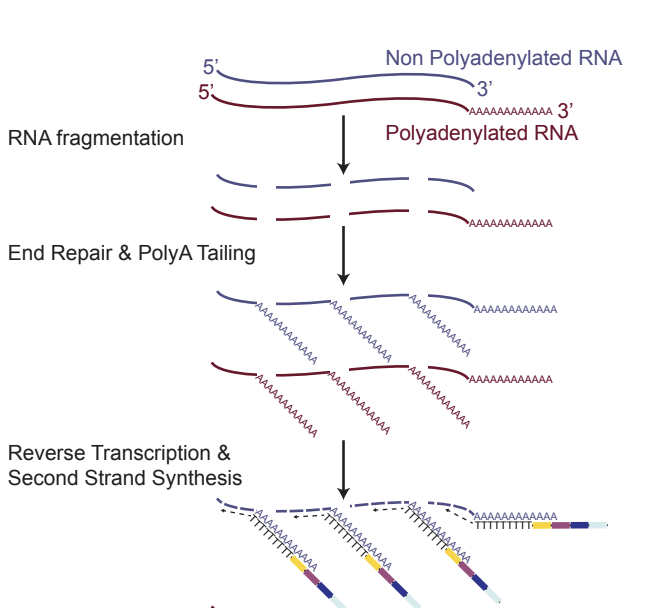
The method fragments the entire RNA payload of a cell and polyadenylates each fragment which makes them compatible with all poly(T) based barcoding methods, enabling UMI-tagging and retaining strand-specificity (2/n) 🧑🔬👨🔬
We @hollfelderlab at @CamBiochem scaled up the process by using droplet #microfluidics to process thousands of cells in a single reaction, with many features adapted from inDrop. Special thanks to our microfluidics guru @TomaszK59462957 for helping me out ! (3/n)
Both droplet and plate-based VASA-seq outperform popular #singlecell #rnaseq methods in terms of capture efficiency, gene body coverage and non-coding RNA representation. This uncovers new layers of information, such as alternative splicing and non-coding markers (4/n)

We then used the droplet method to profile mouse #development, including gastrulation and early organogenesis at stages E6.5, E7.5, E8.5 and E9.5 (~30k cells in total). Thanks to our embryology wizards @t_n_kohler, Ayaka Yanagida and Jennifer Nichols 🧙♂️ (5/n)

@annionna then masterfully analysed and integrated the dataset with an equivalent 10x dataset, and we discovered a whole lot of new cell-type specific markers that will complement current databases! 🖥️ (6/n)

Anna then teamed-up with @AMartinezArias and used this information to define and refine cell-type annotations across timepoints 🐁 (7/n)

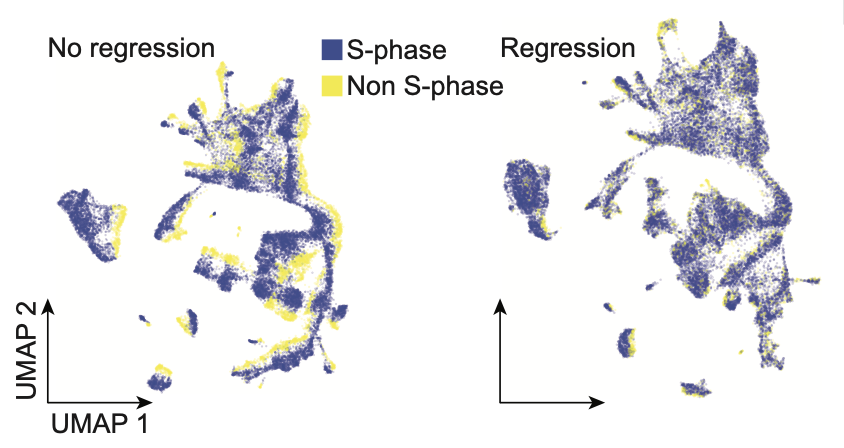
One of the cool features of the method is that it detects a whole lot of histones. This can be used to precisely assign cell-cycle stage, regress out cell-cycle effect and to identify cell-type specific histone usage and cycling pace 🔄 (8/n)
Another feature of the method is the increased capture of unspliced RNA fragments compared to #10xgenomics which boosted #RNAvelocity predictions across developmental trajectories➡️ (9/n)

We then teamed-up with @ge_parada and @m_hemberg to see if the full-length coverage offered by the method uncovered cell-type specific #alternativesplicing motifs, and there was a lot there! This was adapted from their MicroExonator pipeline bit.ly/3ly0XiG 📈(10/n)
We devised two strategies to characterise new splicing patterns across the atlas: differentially included splicing nodes (DISNs) across closely-related cell types, and splice node markers (SNMs) which deviate from the mean 📊 (11/n)

Because blood formation and heart morphogenesis showed extensive splicing remodelling, we dived deeper into these two early organogenesis processes, and for example identified extensive membrane cytoskeletal re-arrangements in primitive erythrocytes 🩸♥️(12/n)

Thank you for all the incredible scientists that enabled this project, which was a big part of my PhD at @Cambridge_Uni. Let us know how you get along with VASA-seq, and looking forward to all the potential applications! (13/n)
• • •
Missing some Tweet in this thread? You can try to
force a refresh


