I recently have claimed, waggishly, that Omicron can:
- Read scientific papers bit.ly/34L0Tro
- Predict the effect of mutations bit.ly/34MhMlu
Pretty smart virus ...
Does this challenge the natural origin hypothesis?
Let's review the weirdness of Omicron
- Read scientific papers bit.ly/34L0Tro
- Predict the effect of mutations bit.ly/34MhMlu
Pretty smart virus ...
Does this challenge the natural origin hypothesis?
Let's review the weirdness of Omicron
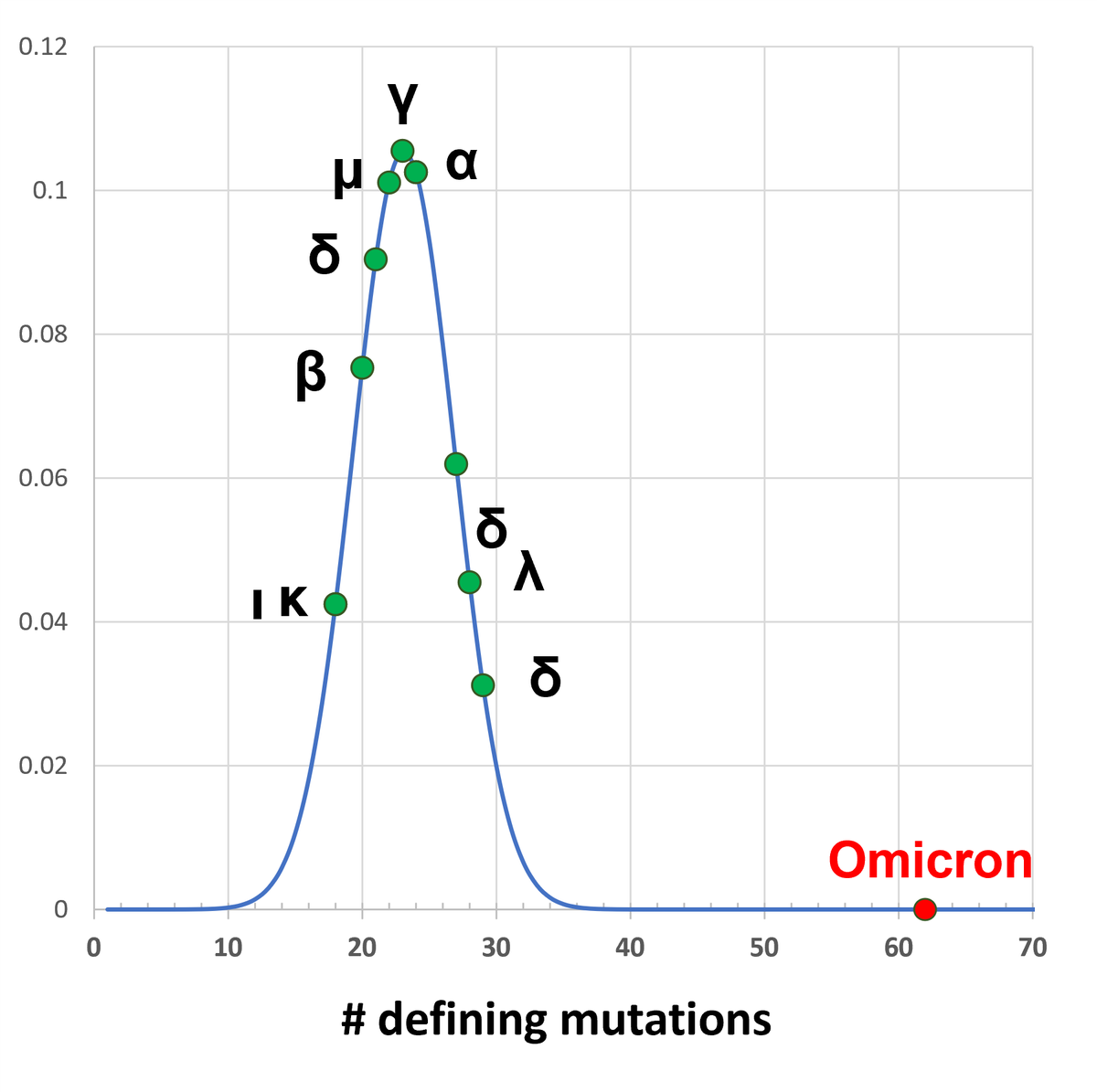
Weirdness #1.
It has 62 new mutations compared to its predecessors.
Previous variants (alpha through mu) have between 18 and 29 mutations.
So, 62 is a lot.
Too many to arise from a natural process?
Probably not. It would just take more time to accumulate that many.
It has 62 new mutations compared to its predecessors.
Previous variants (alpha through mu) have between 18 and 29 mutations.
So, 62 is a lot.
Too many to arise from a natural process?
Probably not. It would just take more time to accumulate that many.

Weirdness #2.
There are no intermediate versions of Omicron that have a subset of the 62 mutations.
Probably the biggest problem for the natural origin idea.
Accumulation takes a long time, and Omicron is more infectious than the measles. How could it hide this long?
There are no intermediate versions of Omicron that have a subset of the 62 mutations.
Probably the biggest problem for the natural origin idea.
Accumulation takes a long time, and Omicron is more infectious than the measles. How could it hide this long?
Weirdness #3.
Mutations come in 2 flavors, compliments of the redundant genetic code:
Silent ("synonymous" or S) mutations do not alter the amino acid they encode
Defining ("nonsynonymous" or NS) mutations do change a protein's AA sequence.
Mutations come in 2 flavors, compliments of the redundant genetic code:
Silent ("synonymous" or S) mutations do not alter the amino acid they encode
Defining ("nonsynonymous" or NS) mutations do change a protein's AA sequence.
Defining (NS) mutations are subject to evolutionary selection pressure, because they tend to alter protein function. Most will be rejected, a lucky few survive. Omicron was lucky 62 times.
With a few exceptions, silent (S) mutations are not selected for or against.
With a few exceptions, silent (S) mutations are not selected for or against.
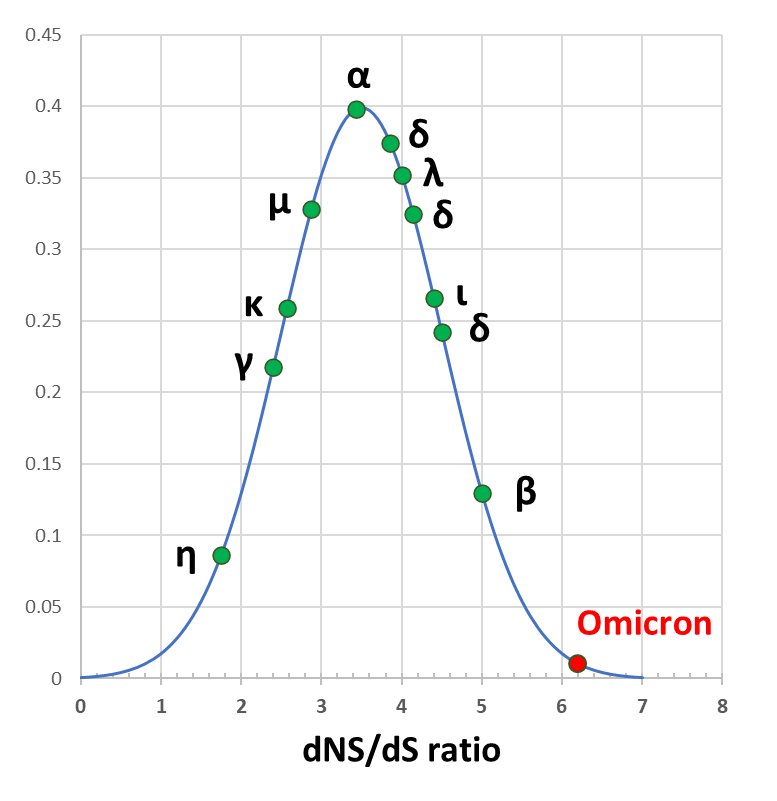
In the absence of evolutionary pressure, the ratio of NS/S can be estimated. I did this for SARS-like genomes and the average ratio is 3.5
Selection pressure can change this ratio.
The spike gene has 30 functional (NS) and 1 silent (S) mutation. Weird:
bit.ly/3FzEpGF
Selection pressure can change this ratio.
The spike gene has 30 functional (NS) and 1 silent (S) mutation. Weird:
bit.ly/3FzEpGF
However, the genome replicates as a single unit, so NS muts in the spike can be compensated by S muts elsewhere. Better to look at the entire genome.
For O NS=62 and S=10. Ratio is 2.6 standard deviations away from the mean: 1% chance to belong to this group. Not impossible.
For O NS=62 and S=10. Ratio is 2.6 standard deviations away from the mean: 1% chance to belong to this group. Not impossible.
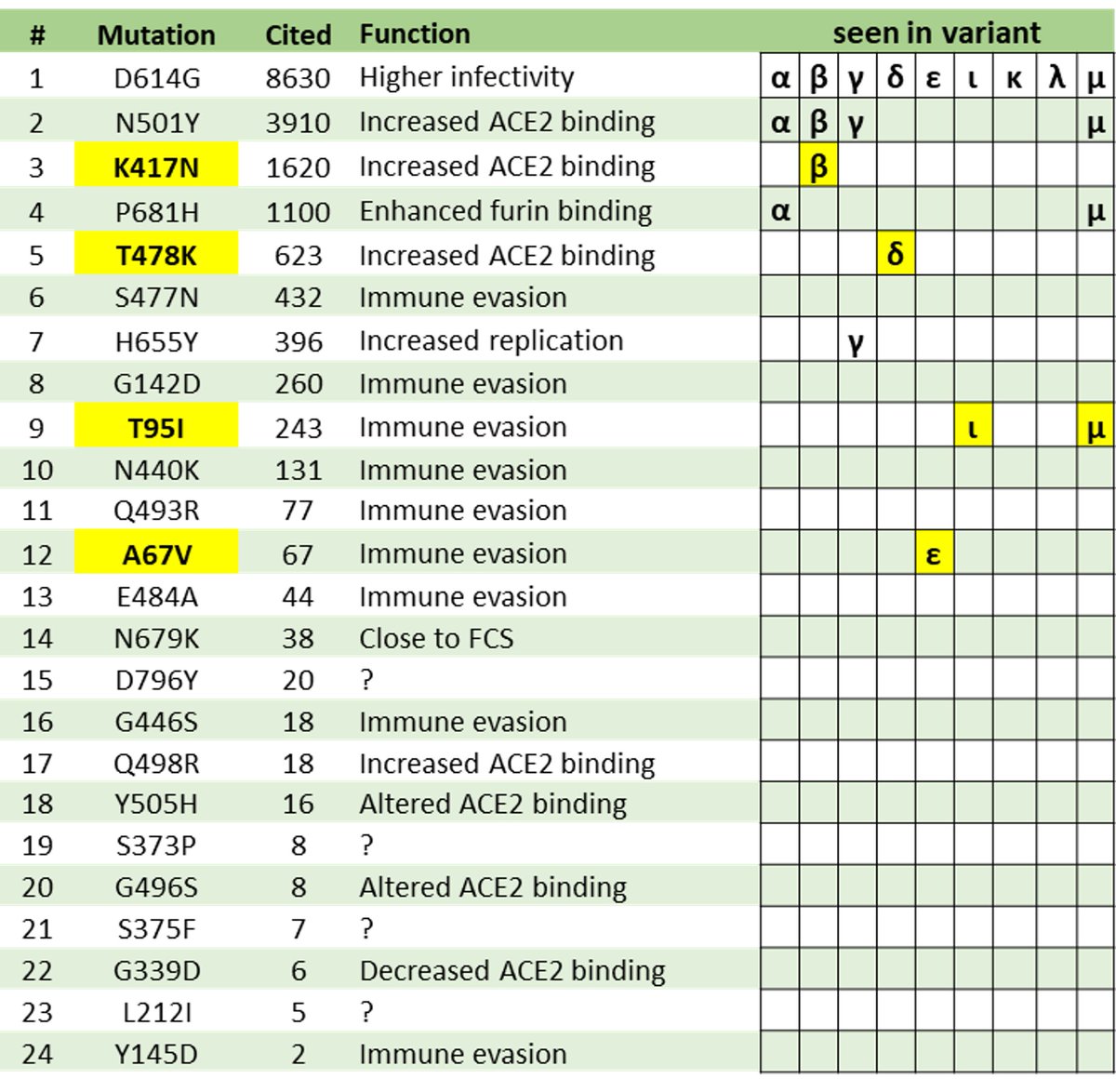
Weirdness #4.
24 of the 30 mutations in the spike of O are published: bit.ly/34L0Tro
Is it therefore designed?
That info could certainly help a designer.
But if a mutation has a clear effect (immune escape, ACE2 binding), evolution can find them as well.
Neutral.
24 of the 30 mutations in the spike of O are published: bit.ly/34L0Tro
Is it therefore designed?
That info could certainly help a designer.
But if a mutation has a clear effect (immune escape, ACE2 binding), evolution can find them as well.
Neutral.
Weirdness #5.
9 Variants of Concern (alpha-mu) predated Omicron.
Of the 30 mutations in Omicron's spike protein, 8 are defining mutations for 7 of its predecessors.
Omicron summarizes everything that came before.
Except it was in hiding all this time.
9 Variants of Concern (alpha-mu) predated Omicron.
Of the 30 mutations in Omicron's spike protein, 8 are defining mutations for 7 of its predecessors.
Omicron summarizes everything that came before.
Except it was in hiding all this time.
That may be a bit more worrisome for the natural hypothesis.
SARS-like viruses can copy gene segments between viruses that co-exist in a person using 'template switching'. But that would imply that Omicron co-existed with 7 of its predecessors at some time during its evolution?
SARS-like viruses can copy gene segments between viruses that co-exist in a person using 'template switching'. But that would imply that Omicron co-existed with 7 of its predecessors at some time during its evolution?
Alternatively, the 8 mutations that O shares with its predecessors are "must have" changes, which increase fitness to such a great extent that evolution will find them, given enough time.
An example of such a mutation is D614G, which is seen in all VoCs.
An example of such a mutation is D614G, which is seen in all VoCs.
Weirdness #6.
There are 12 possible random mutations: from 4 bases (ATGC or AUGC) to the remaining 3. If all have an equal chance, each would happen 8.33% of the time.
But take a look at the list of silent mutations in the SARS2 variants: 63% are C-to-T
There are 12 possible random mutations: from 4 bases (ATGC or AUGC) to the remaining 3. If all have an equal chance, each would happen 8.33% of the time.
But take a look at the list of silent mutations in the SARS2 variants: 63% are C-to-T
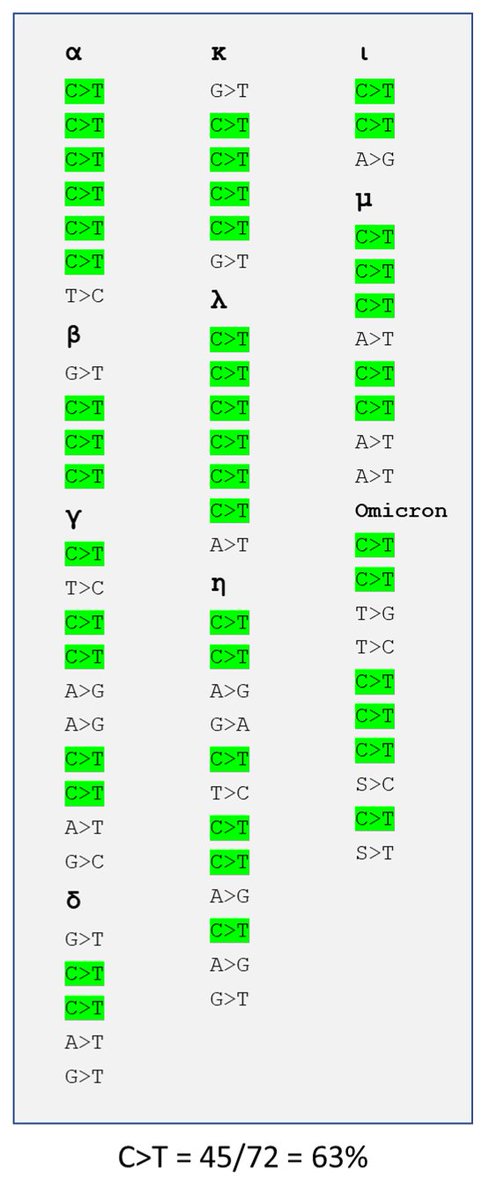
Because these are silent mutations, they are not highly selected. And all variants show the same enrichment for C>T. What gives?
Mutations can arise by 2 mechanisms:
1. Transcription errors
2. RNA editing
APOBECs can convert a Cytidine base to Uracil (C>U or C>T in DNA).
Mutations can arise by 2 mechanisms:
1. Transcription errors
2. RNA editing
APOBECs can convert a Cytidine base to Uracil (C>U or C>T in DNA).
So it appears both mutation mechanisms are at work here, with RNA editing responsible for 63-8.3 = 54.7% of the changes. And because these are silent mutations, they report unbiased about the underlying processes.
So what about defining (NS) mutations?
Glad you asked ...
So what about defining (NS) mutations?
Glad you asked ...
First of all, C>U editing can only account for a very limited number of amino acid changes. See C>U mutation matrix below. Silent mutations in blue. Only 12 NS mutations are possible. Obviously, transcription errors will have to make up for this limited repertoire of RNA editing.
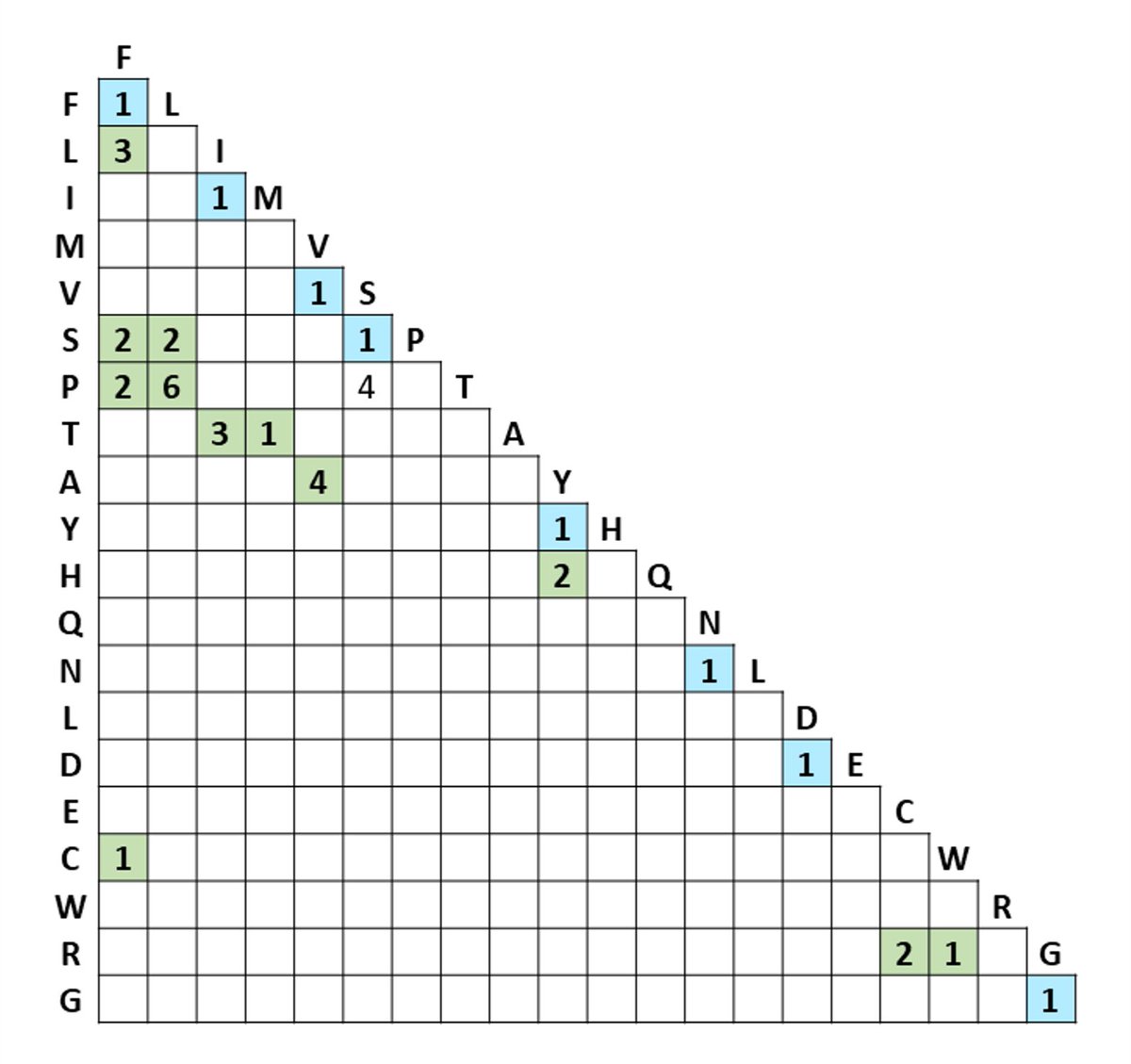
From the silent mutations we know that more than 1/2 of all mutations are generated by RNA editing and result in C>U.
Lets take a look at Omicron below. Of the 30 defining mutations, 5 are C>T (1 is silent in S271L). 17% is much smaller than 53%.
What does that mean?
Lets take a look at Omicron below. Of the 30 defining mutations, 5 are C>T (1 is silent in S271L). 17% is much smaller than 53%.
What does that mean?

RNA C>U editing generates 4 times more defining than silent mutations (see mutation matrix above). So without evolutionary selection, we expect much more than 63% C>T mutations. Instead we see 17%. So selection pressure has removed a very large number of C>U mutations?
• • •
Missing some Tweet in this thread? You can try to
force a refresh