Are you interested in how we can learn more human biology by integrating #singlecell genomics and #GWAS?
Please check out our preprint:
#V2F mapping at single-cell resolution through network propagation
biorxiv.org/content/10.110…
Led by @fulong_yu! A short 🧵 (1/n)
Please check out our preprint:
#V2F mapping at single-cell resolution through network propagation
biorxiv.org/content/10.110…
Led by @fulong_yu! A short 🧵 (1/n)

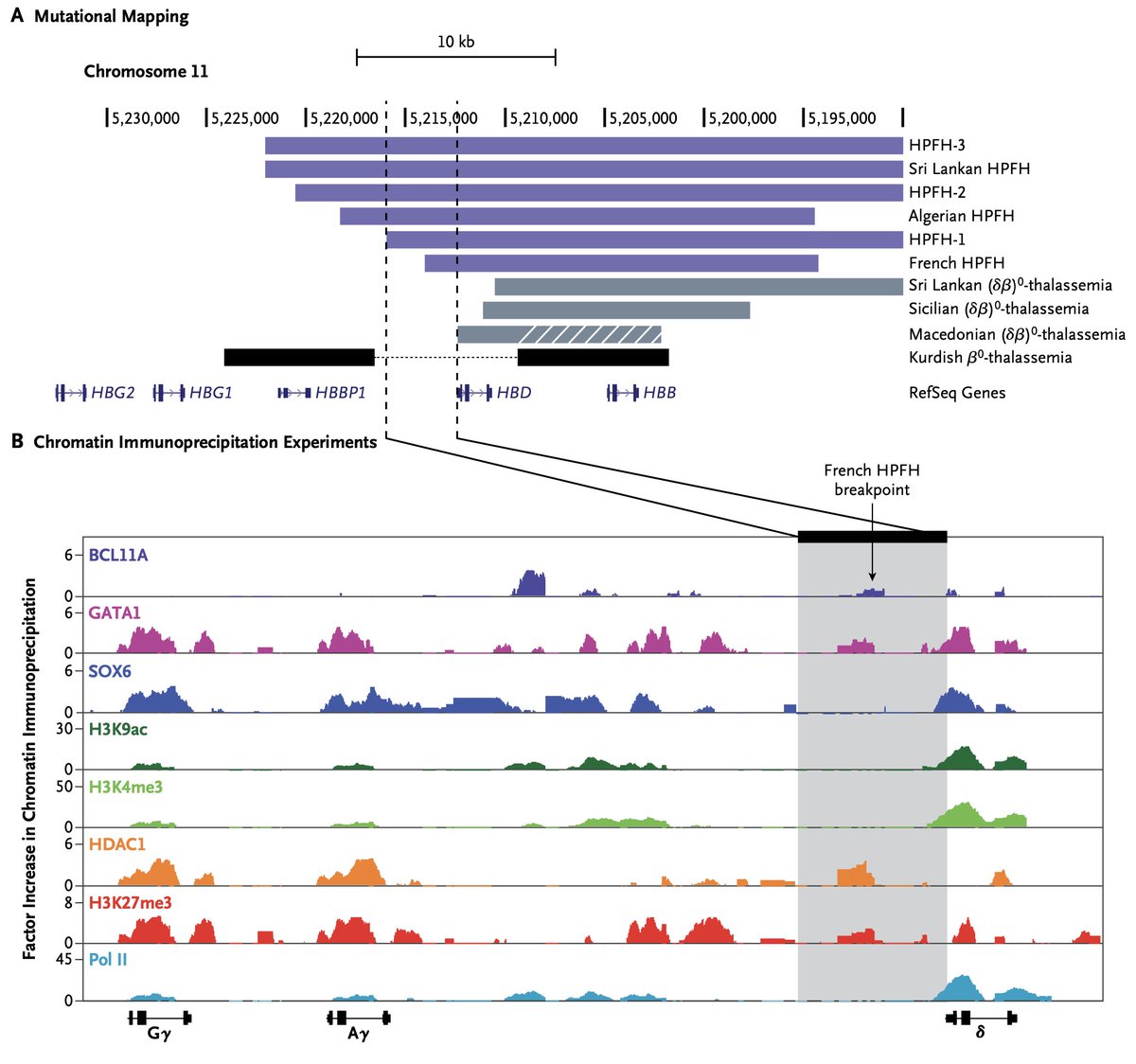
#GWAS have identified innumerable variants associated with disease, as shown in this recent @GWASCatalog plot, yet our understanding of the function of most variants is lacking. #V2F (2/n)

With the increasing availability of #singlecell genomic atlases, such as the @humancellatlas, there are tremendous opportunities to systematically map variants to functional regulatory elements, which can be defined by accessible chromatin or epigenomic marks. (3/n)
But sparsity and noise are major barriers. However, if we use high-dimensional features from such data, we could then in theory better identify enrichments by co-localization or other approaches to define relevant cellular contexts in which #GWAS variants act. (4/n)

.@fulong_yu realized that network propagation methods, as @Google used in their #PageRank algorithm, could enable this. (5/n)

He developed a method we call SCAVENGE (Single Cell Analysis of Variant Enrichment through Network propagation of GEnomic data) to enable this. It outputs a trait relevance score (TRS) for all the cells that one studies after network propagation. (6/n)

We initially tested this with simulated #singlecell data with a model complex trait: genetic variants associated with monocyte count. This worked quite well and enabled better separation than simply examining co-localization metrics. (7/n)
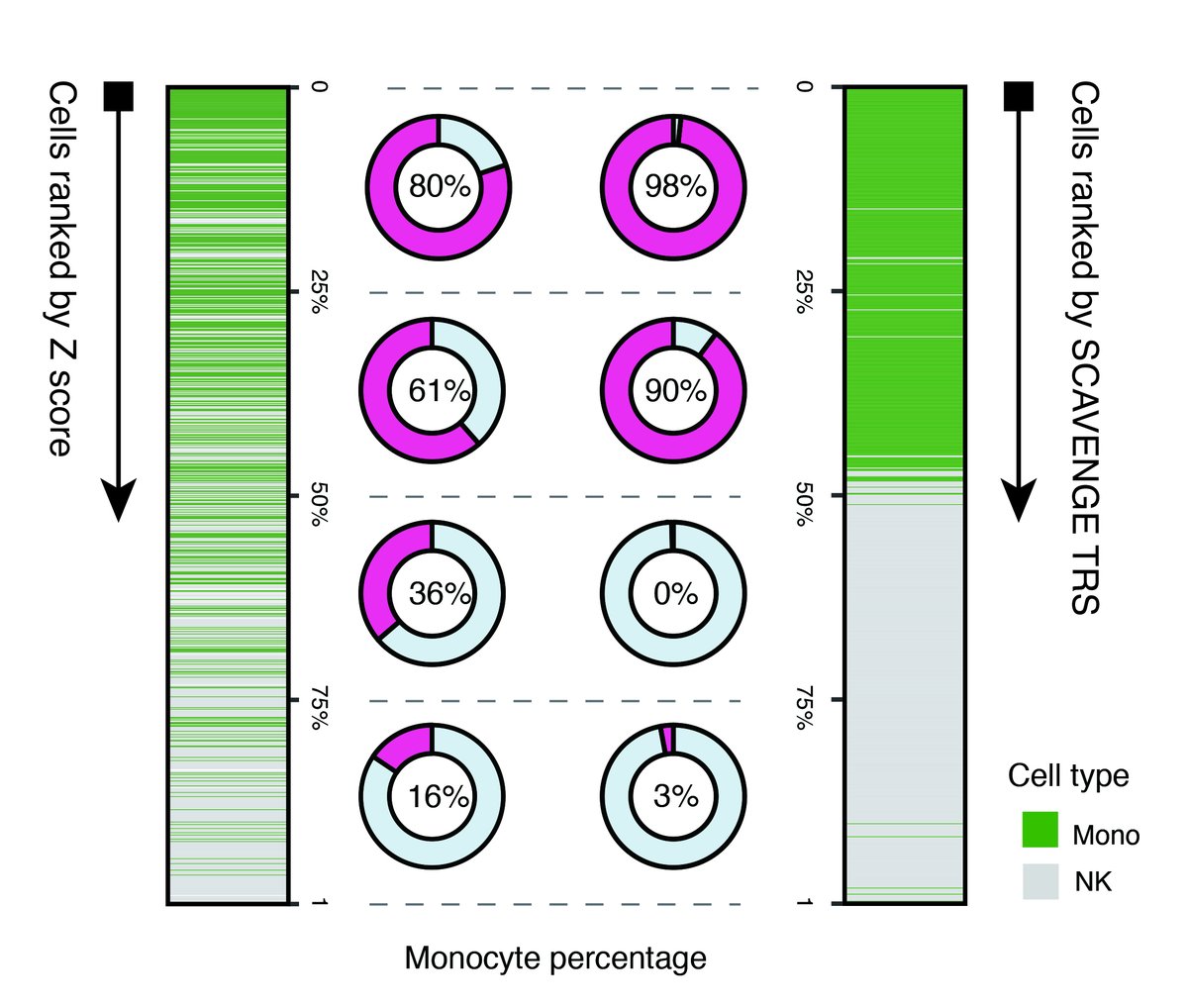
We then examined real #singlecell data from peripheral blood mononuclear cells (PBMCs) and could see that SCAVENGE enabled robust separation of relevant cells (monocytes) for this trait of interest as well. (8/n)

We could also study a range of hematopoietic phenotypes and show that we get robust enrichments across #singlecell ATAC-seq analyses from human bone marrow, as we had observed in prior studies in bulk cells, but with additional enrichments that we had missed at lower res. (9/n)

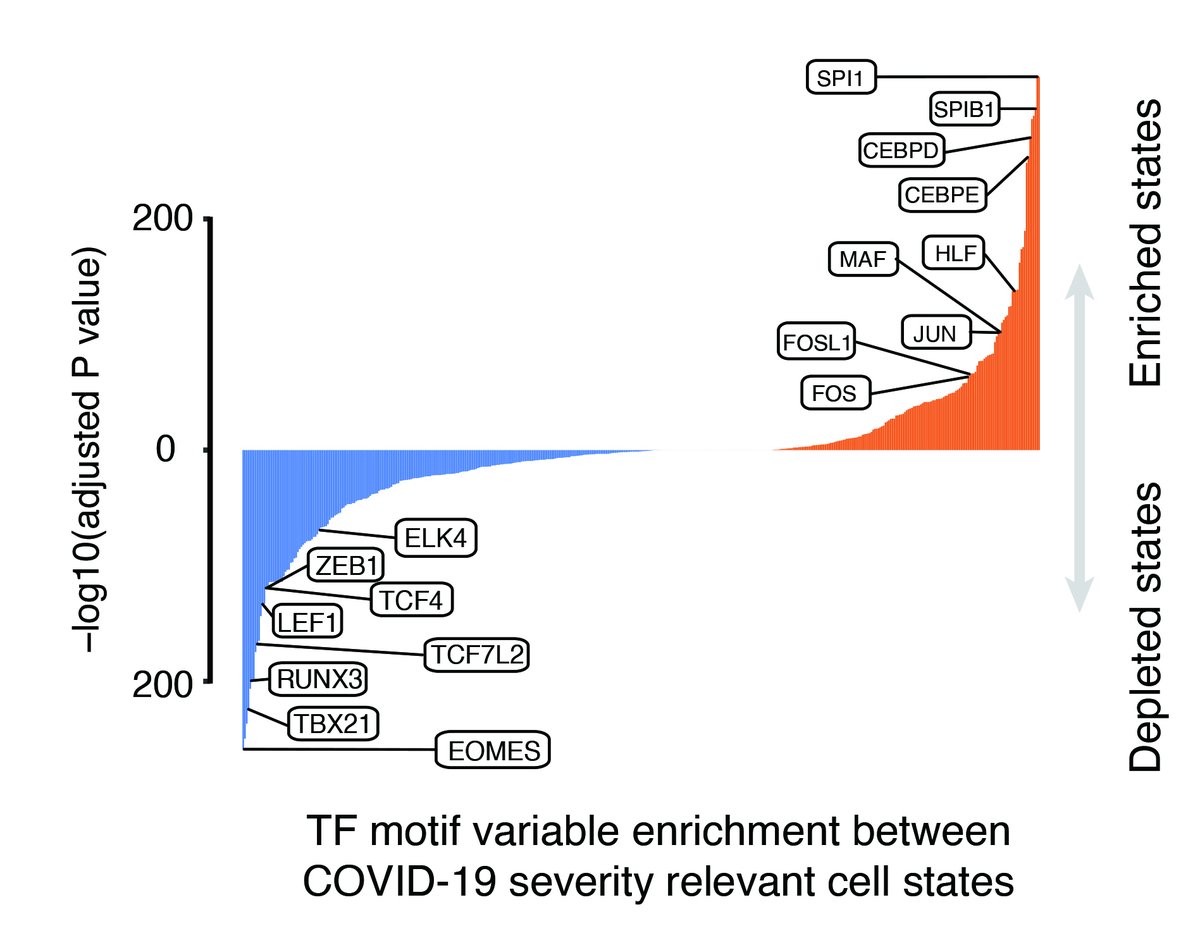
We next applied SCAVENGE to PBMC scATAC-seq data from individuals w/ #COVID19 and healthy controls. This revealed strong enrichment of #GWAS variants from @covid19_hgi in monocytes and dendritic cells. However, we also noted heterogeneous enrichments. (10/n)

When we examined this further, we realized that the CD14+ monocytes that are typically grouped together had a set of trait enriched cells that were expanded in severe #COVID19 infections and appeared to be a more immature subset driven by SPI1 and other TFs. (11/n)

Finally, we wanted to see how well SCAVENGE could perform across a cell trajectory. We examined B lymphocyte differentiation for enrichment of childhood acute lymphoblastic leukemia (ALL) #GWAS risk variants... The cell-of-origin in ALL is unknown. (12/n)

Our analysis suggests a strong enrichment in pre-B cells, as nicely illuminated in our analysis of a risk variant at the CEBPE locus. (13/n)

We also observed strong enrichments of PAX5 motifs that correlated with the TRS for ALL risk. Germline PAX5 variants predispose to ALL and our data suggests that cis-regulatory elements interacting with PAX5 may also confer risk for ALL. (14/n)

Please check out the pre-print. SCAVENGE is available as an R package: github.com/sankaranlab/SC… (15/n)
This work was pioneered by the amazing @fulong_yu with valuable assistance from @liamcato, @chenweng1991, @la_liggett, @kerenxuepi, @JswLab, @adamdesmith, and other friends and colleagues. (fin)
• • •
Missing some Tweet in this thread? You can try to
force a refresh