Organization and expression of the mammalian mitochondrial genome go.nature.com/3MqLEV0 #Review by @TheRackhamLab & @FilipovskaLab
The mitochondrial genome is co-localized with DNA replication machinery, which maintains and propagates it in cells, and gene expression machinery, which synthesizes essential mtDNA-encoded components of the respiratory chain required for energy conversion 
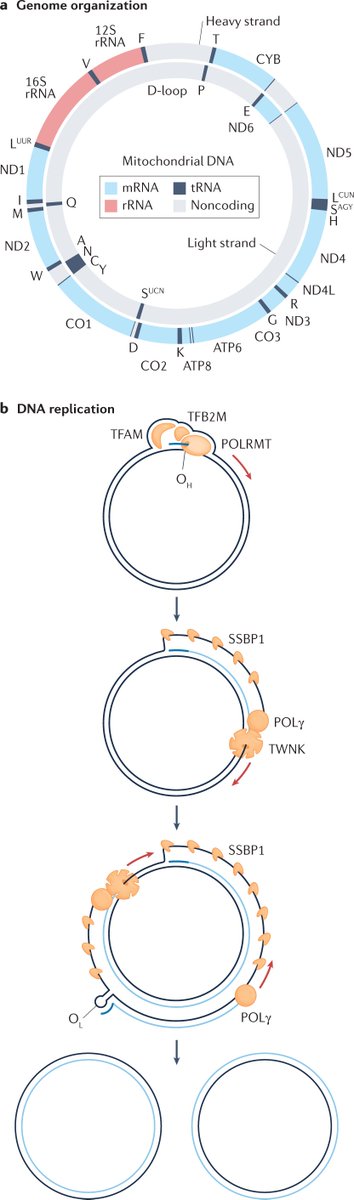
Mammalian mitochondria produce a minimalistic complement of 11 mRNAs, 22 tRNAs, and two rRNAs, however, their metabolism is predominantly regulated post-transcriptionally by nuclear-encoded RNA-binding proteins during RNA processing, maturation, translation and decay

Advances in high-throughput sequencing technologies and cryoelectron microscopy have shed light on the structure and organization of the mitochondrial genome and revealed unique mechanisms of mitochondrial gene regulation
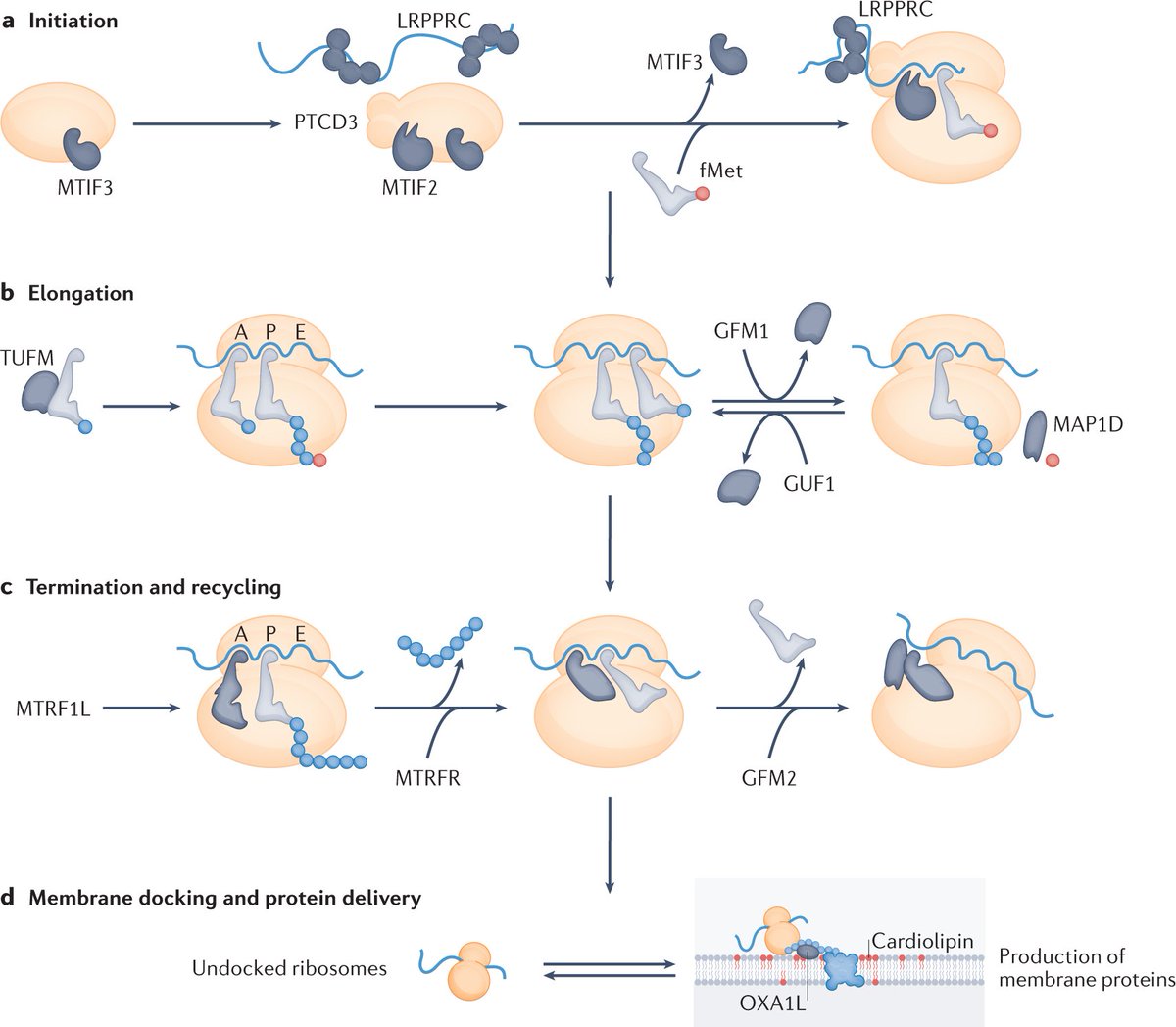
New animal models of impaired mitochondrial protein synthesis have shown how the coordinated regulation of the cytoplasmic and mitochondrial translation machineries ensures the correct assembly of the respiratory chain complexes
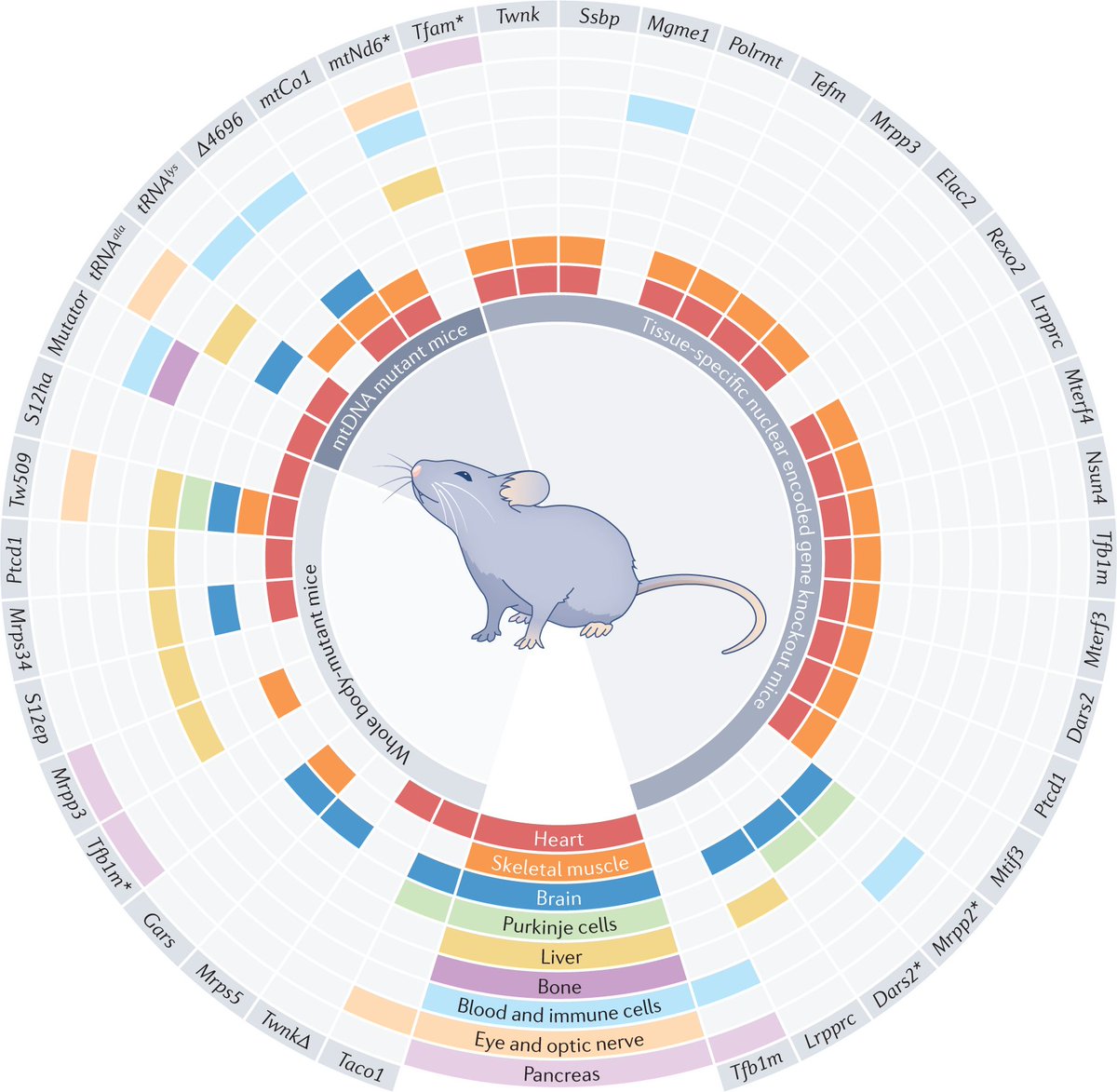
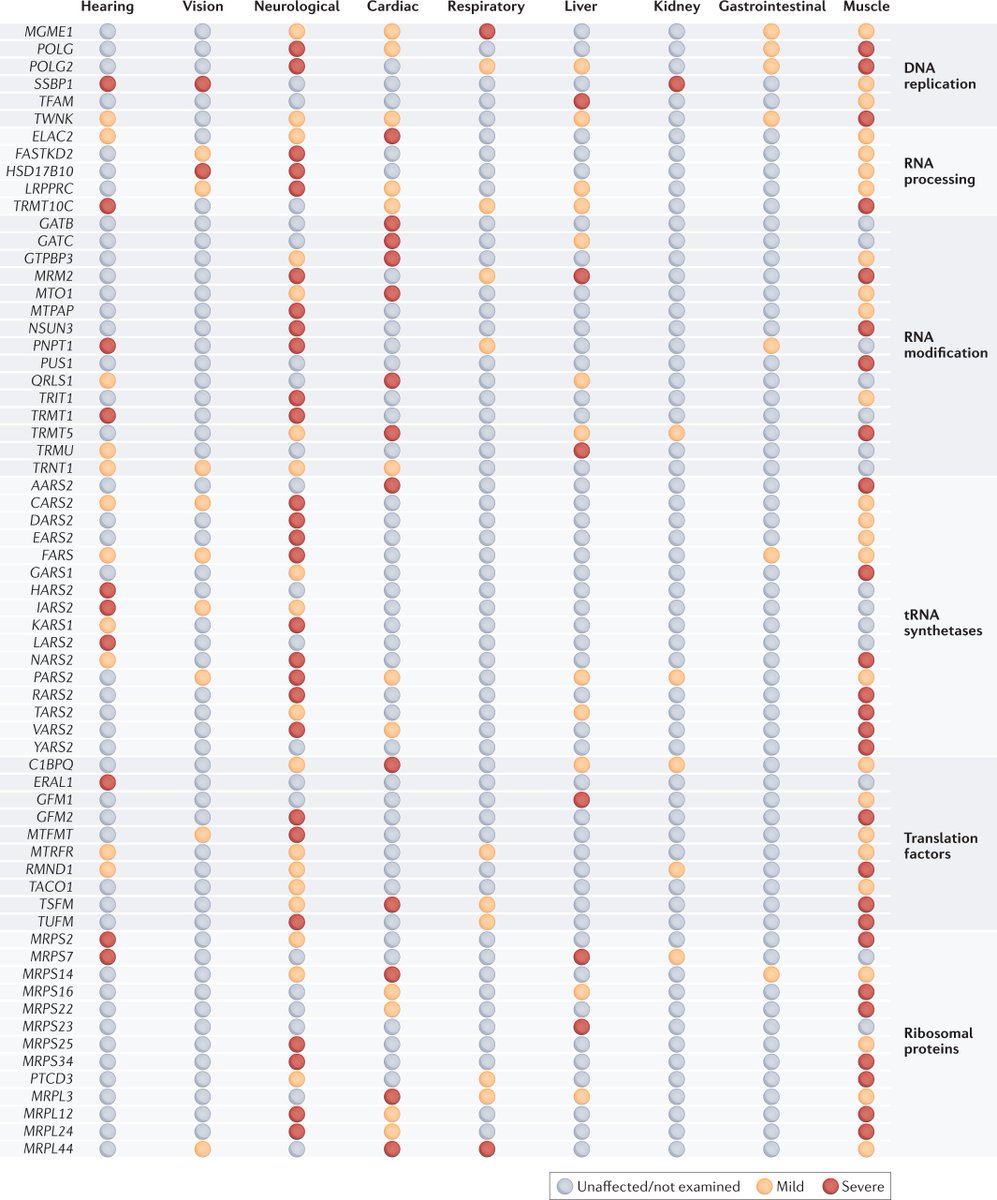
Defects in mitochondrial genome regulation and expression are responsible for mitochondrial diseases, and also contribute to common diseases such as cardiovascular, metabolic and neurodegenerative diseases, cancer and ageing
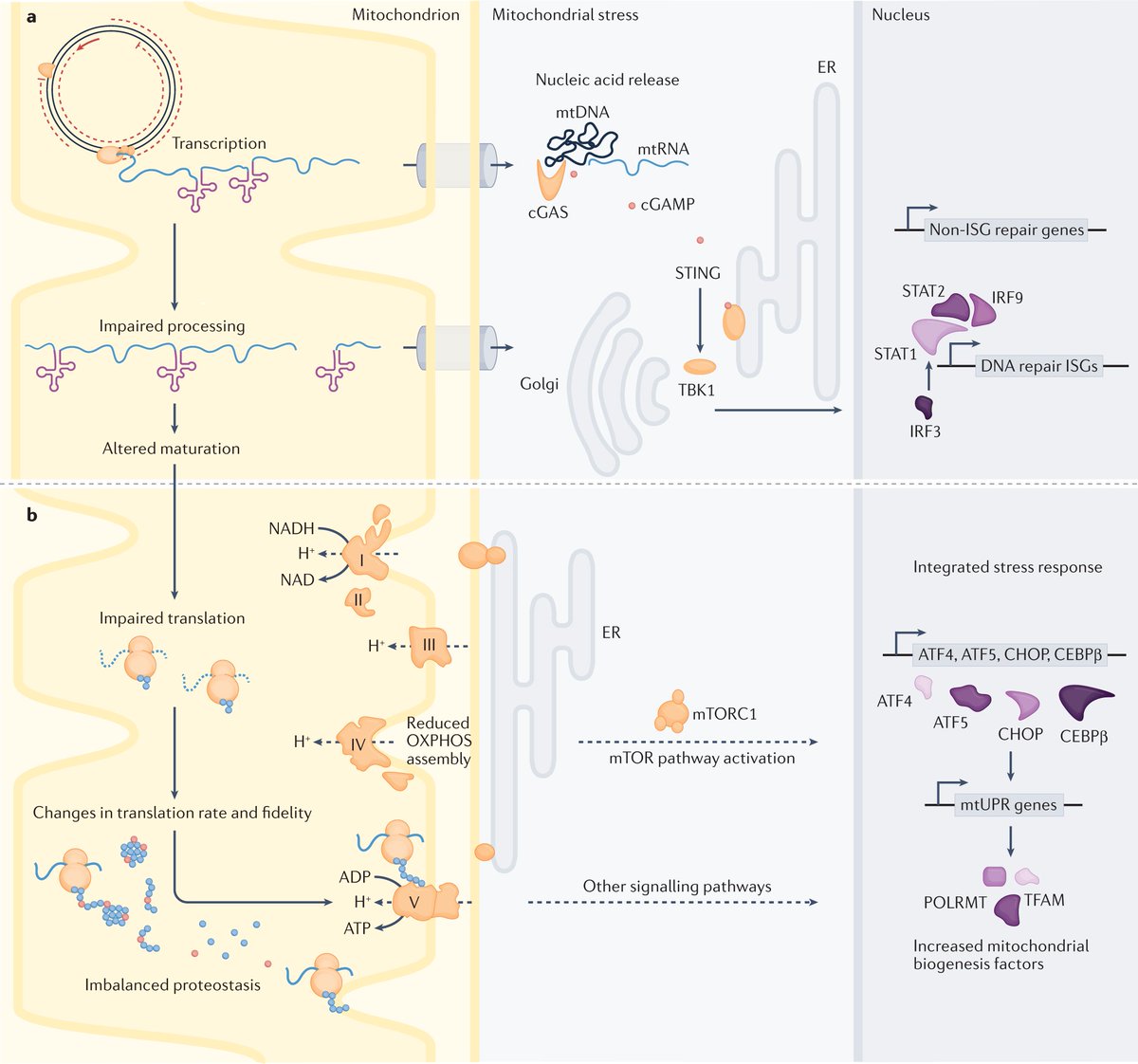
Here, Rackham and Filipovska review our current understanding of mitochondrial genome organization and expression and showcase studies that dissect how mitochondrial signals are communicated to the rest of the cell to trigger responses to altered mitochondrial gene expression
Free to read here: rdcu.be/cL3jF
• • •
Missing some Tweet in this thread? You can try to
force a refresh