The analysis of single-cell RNA-seq data begins with "normalizing" counts. In a preprint with @sinabooeshaghi, @IngileifBryndis & @agalvezmerchan, we examine the assumptions and challenges of normalization, benchmark methods, and motivate solutions: biorxiv.org/content/10.110… 🧵 1/
We weren't particularly interested in studying normalization, but faced a vexing problem related to normalizing feature barcodes. In scouring the literature for solutions to our problem, we became increasingly confused rather than enlightened about how to normalize our data. 2/
We started with the excellent recent review / expository article by @const_ae & @wolfgangkhuber that looks at strengths & weaknesses of many methods: biorxiv.org/content/10.110…. It became clear to us that a central question is how to normalize depth w/ gene count overdispersion. 3/
An important publication focusing on this question is the sctransform paper written by Hafemeister & Satija (from the Seurat team) in 2019:
genomebiology.biomedcentral.com/articles/10.11…. They proposed using Pearson residuals derived from regularized negative binomial regression. 4/
genomebiology.biomedcentral.com/articles/10.11…. They proposed using Pearson residuals derived from regularized negative binomial regression. 4/
The term "sctransform" is both a statistical method, as described in the Hafemeister & Satija paper, but it's also software implemented as part of Seurat. In trying to understand exactly what the two are, and how they relate to each other, we descended into a squirrel hole.. 5/ 

Starting with the software, it became a problem that Seurat is not published, and there is no explanation anywhere of exactly how normalization is implemented in the program or how it's used in the various Seurat functions. So we made a map for ourselves by reading the code. 6/
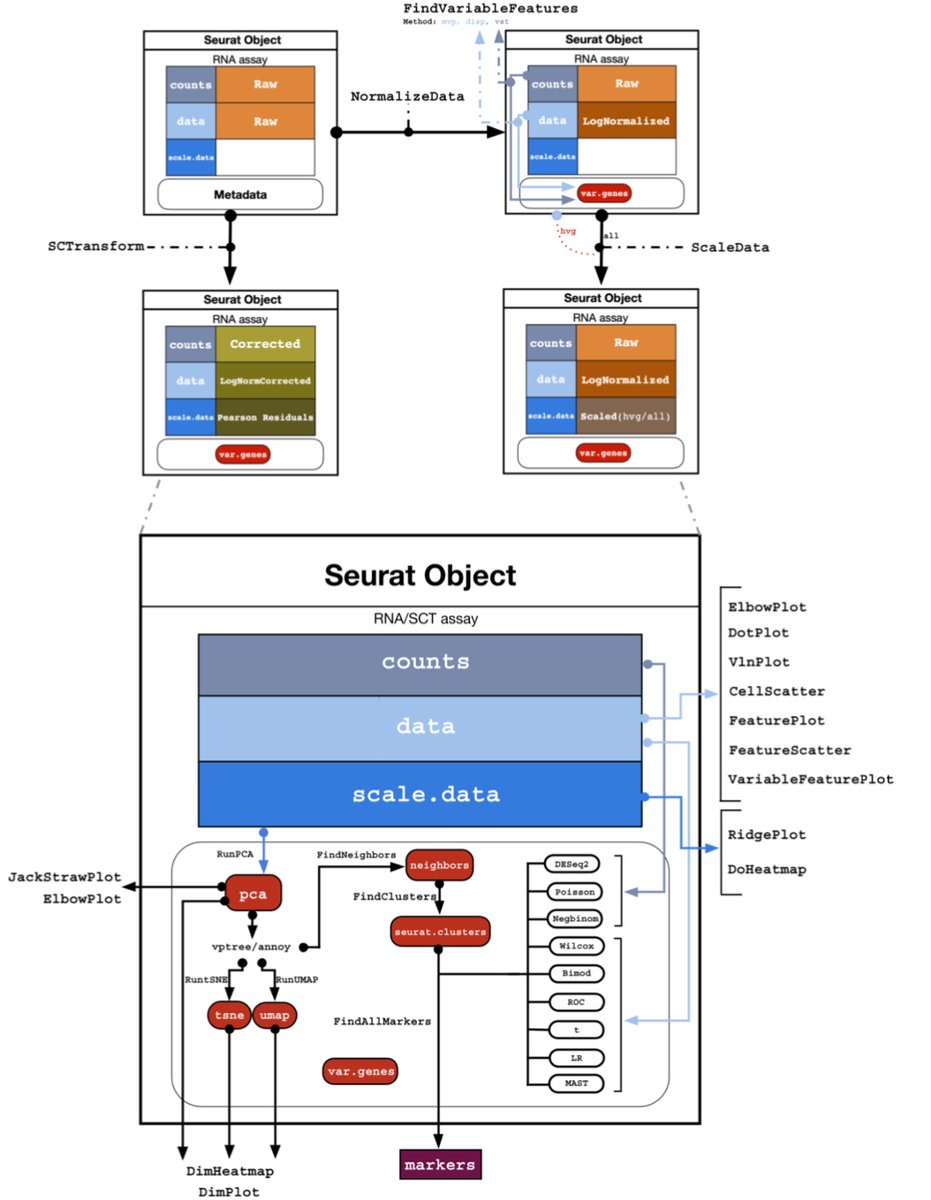
A lot of the normalization literature focuses on statistical aspects of various transformations, but the reality of Seurat is that results are much more influenced by software engineering decisions; different normalizations interact with each other in non-trivial ways. 7/
By the way, it's a similar story in Scanpy, but a full comparative analysis & explanation is a matter for another 🧵 I will say, as have others, that it's a serious problem for the field with implications for many frequently performed analyses. 8/
https://twitter.com/davisjmcc/status/1524178348863520770?s=20&t=KlkFnIEQ320hQbXnw5c9ew
The engineering matters but is not to say statistical details are not important. As @JanLause, @CellTypist and @hippopedoid have shown, the sctransform gene-specific overdispersion estimates are not only superfluous but possibly detrimental. genomebiology.biomedcentral.com/articles/10.11… 9/
But the reality of #scRNAseq normalization right now is that in packages such as Scanpy and Seurat, choosing a "method" means choosing a collection of methods, not a single one. We asked whether there can be a single transformation that could serve multiple tasks. Tl;dr yes! 10/
To clarify what properties a transformation should have, we surveyed the needs of several common tasks (e.g., dim. reduction, differential expression, marker gene finding etc.) BTW we're not the first to do this, see e.g. nature.com/articles/nmeth… by @CataVallejosM et al. 11/
But we took a nuanced view on some tasks. E.g., we distinguish differential expression from finding markers, two tasks which are unfortunately frequently confounded. A good marker is not only higher (in expression) in a cell type relative to others, it is also specific. 12/
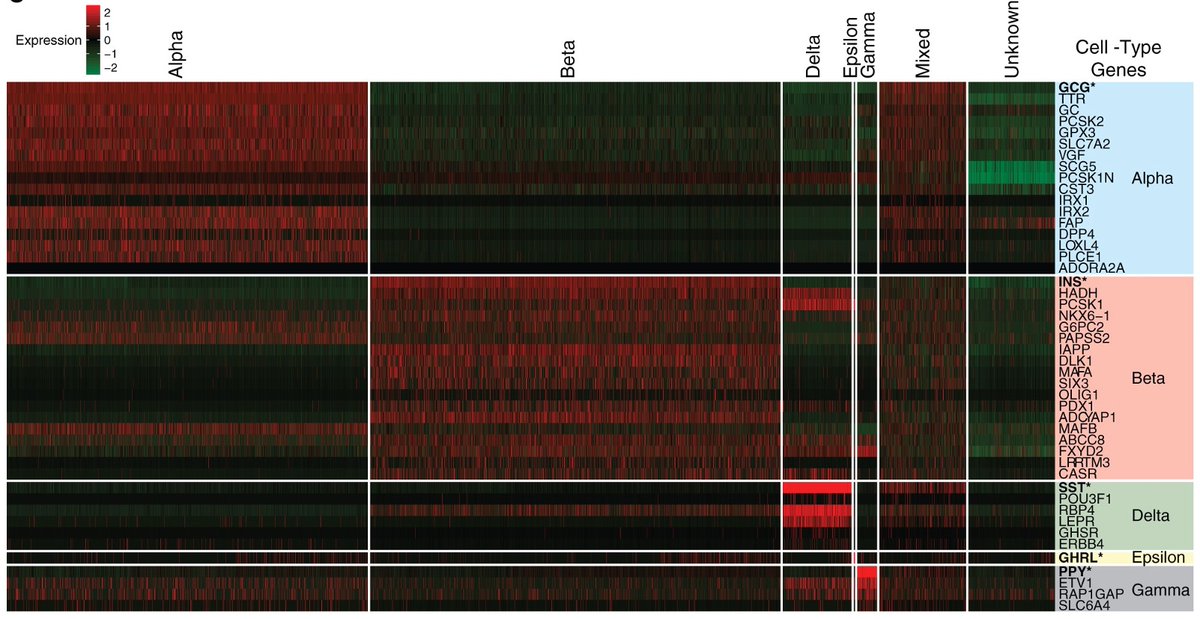
Consider, e.g., the heatmap in @LBCastle et al. from the @NIHDirector (Francis Collins') lab. Markers require diff. abundance not just across cell types, but also genes within cell types (rows & columns). This calls for monotonic normalization, otherwise cols. are scrambled. 13/
We concluded that three properties of normalization are key: variance stabilization, depth normalization, and monotonicity. Details of how these are relevant for different analysis tasks are in our preprint. We benchmarked several popular methods with respect to these. 14/
The word "popular" here is carrying a lot. There are tons of normalization methods for #scRNAseq that have been developed, including some we think are very interesting, e.g. Sanity by @jeremiebreda, @ZavolanLab and @NimwegenLab. 15/ nature.com/articles/s4158…
But we limited ourselves, for now, to the main methods implemented in Seurat, Scanpy and scprep. Figuring out how users actually use these packages was a Herculean task in and of itself. Only Scanpy from the @fabian_theis lab is published. genomebiology.biomedcentral.com/articles/10.11… 16/
Seurat, which is probably the most popular package for #scRNAseq, has different normalizations recommended in different versions, and multiple versions are in use because R users are reluctant to update R just to switch to a newer version.
https://twitter.com/lobrowR/status/1180121094189023233?s=20&t=Iyu1bz8kZOjvc-Oa2Li3xQ17/
Now to the fun part... we benchmarked on 526 single-cell RNA-seq datasets comprised of ~140 billion reads of data. The uniform processing and analysis of this data was done by two students in my group: @agalvezmerchan and @sinabooeshaghi. 18/
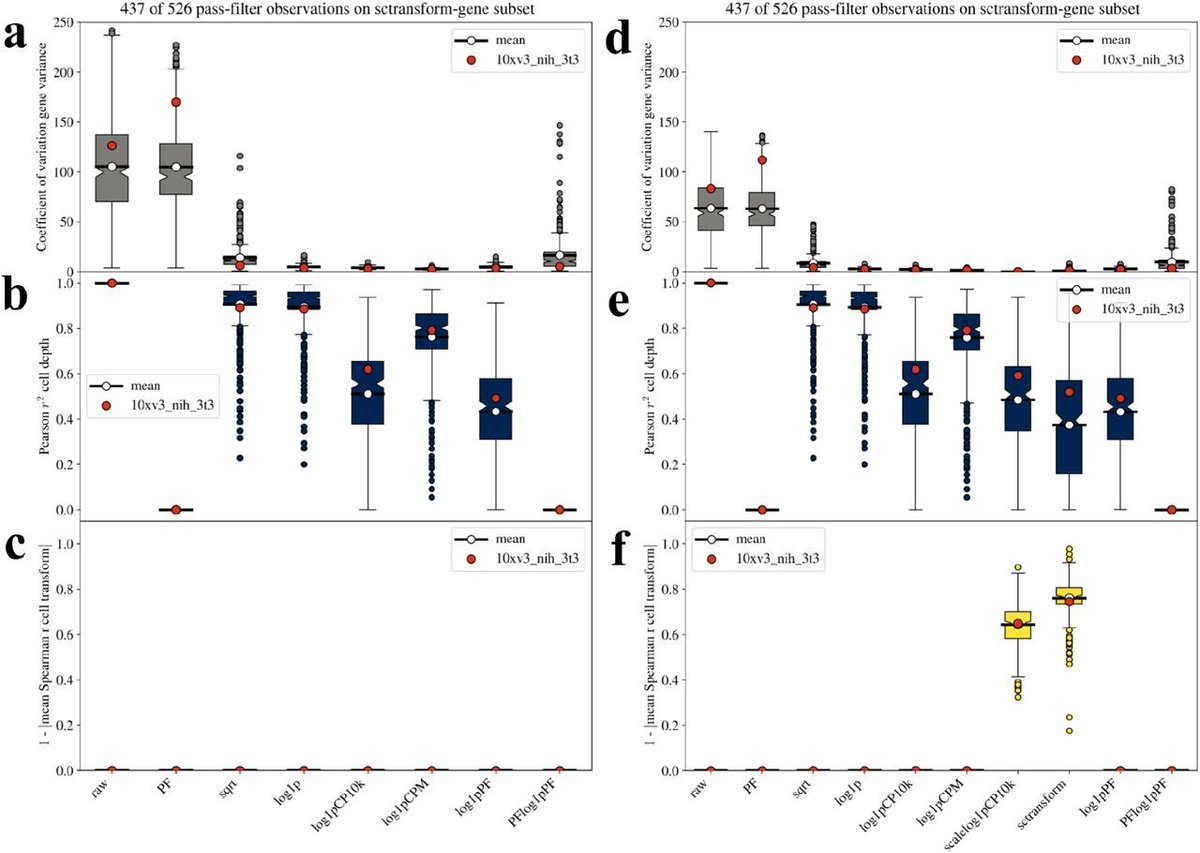
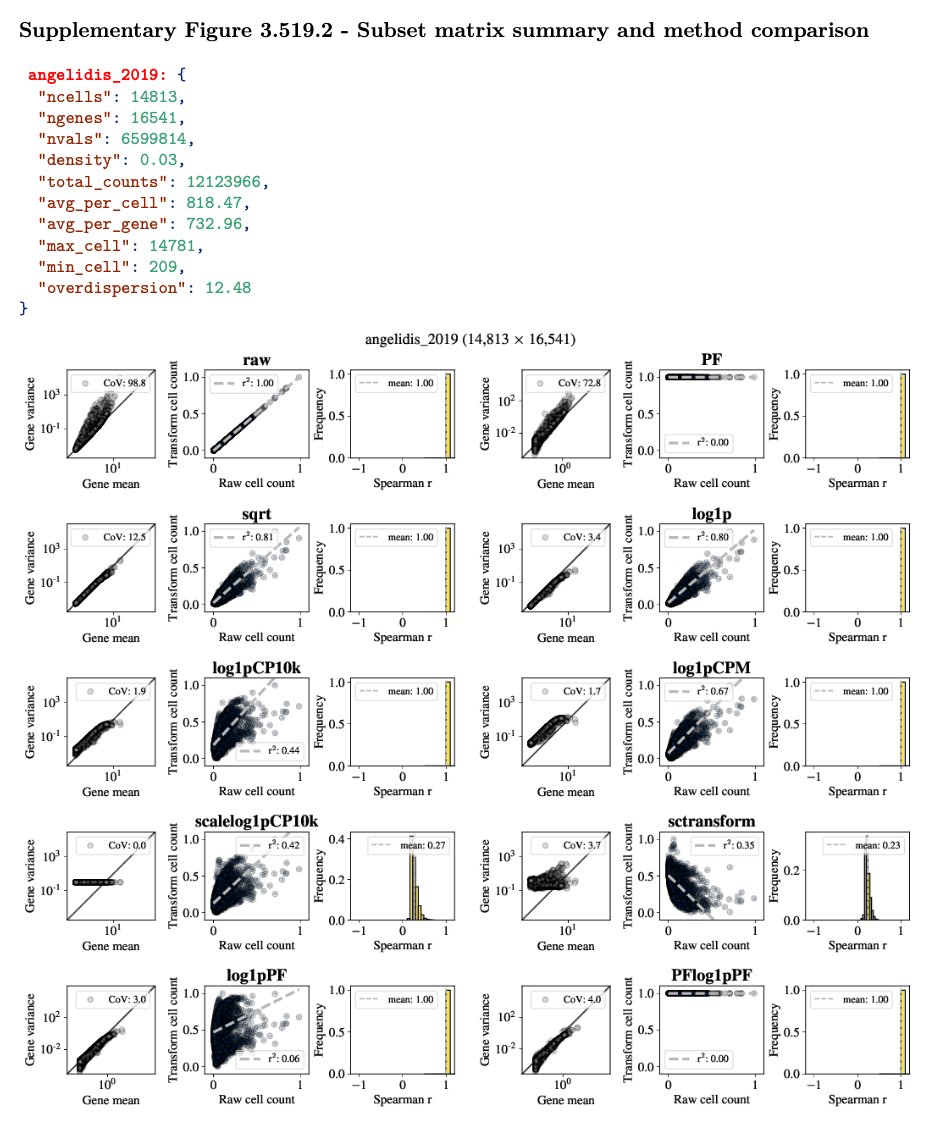
The figure in the previous tweet is mind-boggling to ponder. It summarizes the results for this massive analysis; individual dataset benchmarks are in our supplement which amounts to 1596 pages (!) as a result. An example from a single one of these datasets is shown below. 19/
How @agalvezmerchan and @sinabooeshaghi automated all of this is a matter for another 🧵, because it involved the development of many tools and numerous ideas. As an example, see ffq:
https://twitter.com/lpachter/status/1522322188493197312?s=20&t=ckG6lwqqlbdeg95D9t1t2w20/
What did we learn?
1. Benchmarking on a handful of datasets, as has been the practice to date, is insufficient.
2. The statistical details of methods matter, but even suboptimal transforms (e.g. sqrt) are fine.
3. The PFlog1pPF works well as an overall single transform. 21/
1. Benchmarking on a handful of datasets, as has been the practice to date, is insufficient.
2. The statistical details of methods matter, but even suboptimal transforms (e.g. sqrt) are fine.
3. The PFlog1pPF works well as an overall single transform. 21/
What is PFlog1pPF? ideally normalization procedures should be customized to tasks, but practically it's useful to have a single normalized count matrix for many applications. PFlog1pPF stands for proportional fitting followed by log1p followed by PF; details in the preprint. 22/
There's much more in the preprint. E.g., the very premise of normalization, namely that depth should/must be normalized and variance stabilized is itself problematic, as transforms don't distinguish technical from biological "noise". We will also return to this in a future 🧵23/
The supplement also has many results that may be of practical interest to users. E.g., it turns out that the results of sctransform in practice don't differ much from (the much faster to compute) scalelog1pCP10k. 24/
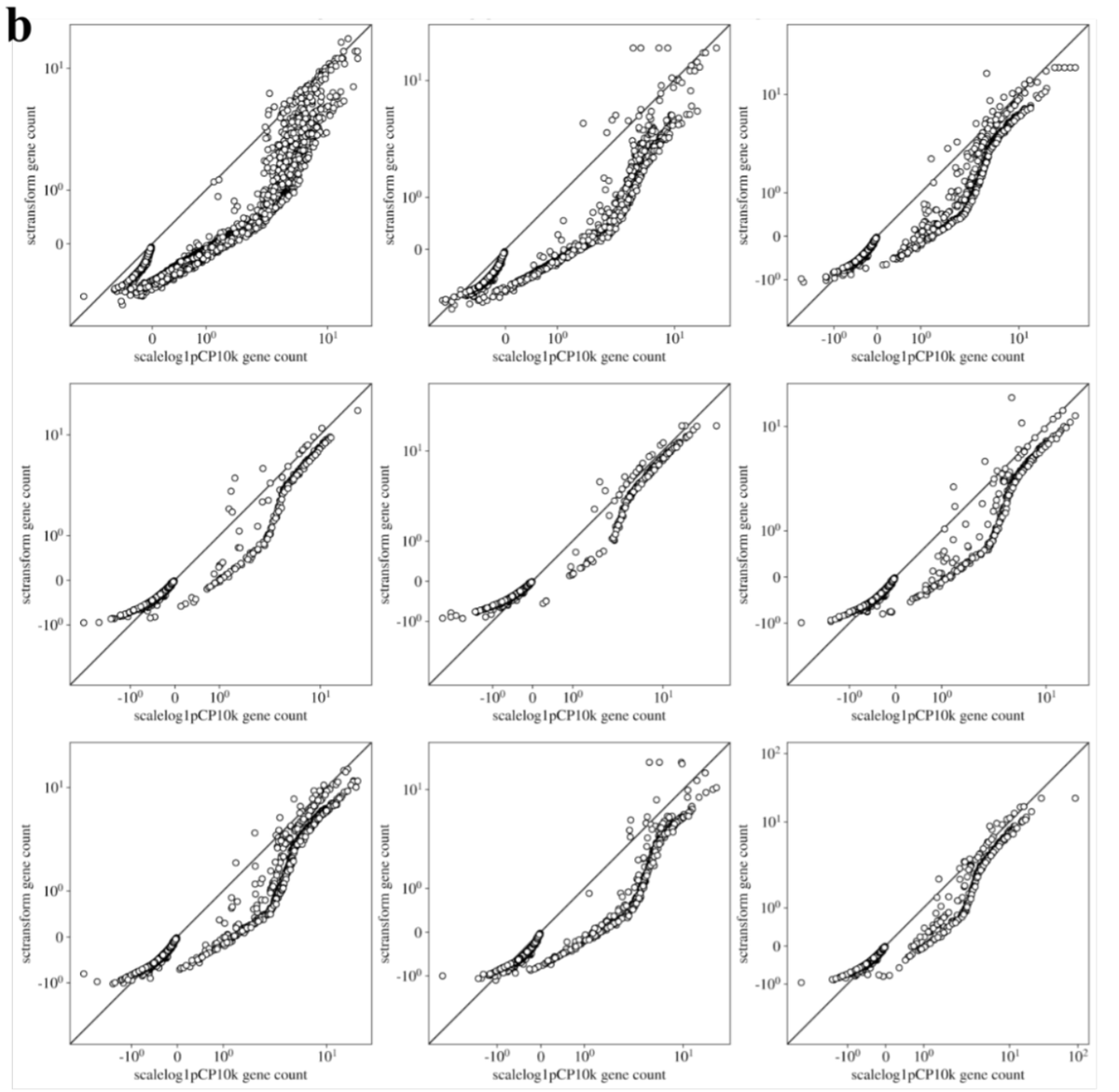
In summary, we hope that our uniformly processed datasets are useful for future work on normalization (available here github.com/pachterlab/BHG…), and that our observations on method performance / software engineering considerations are helpful for practitioners of #scRNAseq. 25/25.
• • •
Missing some Tweet in this thread? You can try to
force a refresh