The exciting reveal of Ultima Genomics last week was accompanied by the publication of four preprints. Intrigued by the potential of the technology, @sinabooeshaghi & I decided to take a look at the data. A 🧵 about our findings & a preprint we posted: biorxiv.org/content/10.110… 1/
We first looked at the company's own preprint on which the CEO is first author: biorxiv.org/content/10.110…
Unfortunately, no data. No code. There is not even supplementary material, which the authors write "will be made available in the near future." 2/
Unfortunately, no data. No code. There is not even supplementary material, which the authors write "will be made available in the near future." 2/
Without data or code, obviously one cannot check the claims of the company. But in this case one cannot even understand the claims. E.g. the description for Fig. 2e in the Methods is useless without code to explain what was actually done to produce it. 3/ 


So we looked at preprint #2, which is on whole-genome methylation sequencing from the Snyder lab: biorxiv.org/content/10.110…
This time: "The datasets used and/or analyzed during the current study are available from the corresponding author upon request." 4/
This time: "The datasets used and/or analyzed during the current study are available from the corresponding author upon request." 4/
I'm tired of this "data available upon request" thing, and I'm not the only one. In the past year I've had to make at least a dozen such requests, and my success rate in obtaining data is even lower than NIH funding rates. 5/
https://twitter.com/ceptional/status/1533567322736435200
So we looked at preprint #3, which published Perturb-seq, some of it done with Ultima: biorxiv.org/content/10.110…
Went to look for the data link in the preprint but only got "Raw sequencing data will be deposited into SRA."
6/
Went to look for the data link in the preprint but only got "Raw sequencing data will be deposited into SRA."
6/
Protip: when reading a @biorxivpreprint preprint it can pay off to chase the tweet links from the @biorxivpreprint page. The authors of this paper did eventually tweet out a link to some of their data (I leave the task of finding it as an exercise to the reader!) 7/
We did eventually find data linked from a preprint... yay preprint #4 on single-cell RNA-seq!! biorxiv.org/content/10.110…
This was the only preprint with an accession (GSE197452) AND code (github.com/seanken/Compar…) AND Supp. material. So we went with data from this preprint.
8/
This was the only preprint with an accession (GSE197452) AND code (github.com/seanken/Compar…) AND Supp. material. So we went with data from this preprint.
8/
Our curiosity was piqued when we read in the abstract that "[Ultima Genomics data]] show comparable results to existing [Illumina] technology" but saw a different story in Extended Data Figure 1: 9/
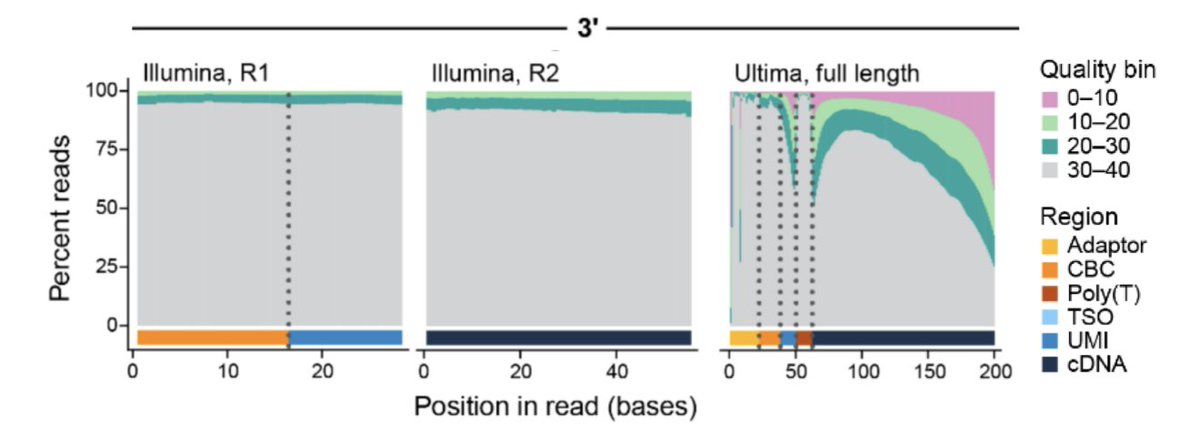
So we decided to analyze the data ourselves to see what's going on. Ultima data is single-end, and pre-processing it required a tool that can handle the placement of the barcodes, UMIs & cDNA. Modifying kallisto | bustools was easy (thanks modularity!) and we now have a tool. 10/
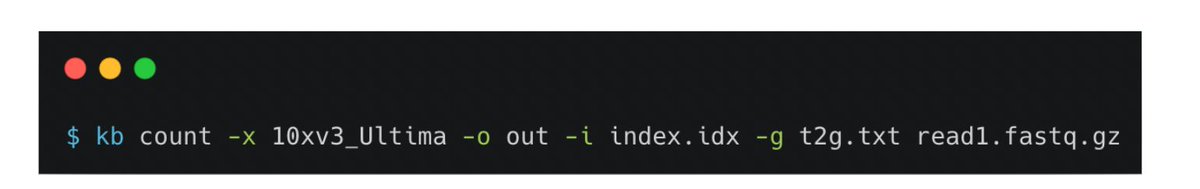
Ok, so now we were ready to do our own apples-to-apples comparison, except the Illumina data had 55bp of cDNA, vs. 174bp in the Ultima (not clear why the authors didn't just sequence standard 150bp Illumina reads). So we trimmed the Ultima cDNA data to 55bp. 11/

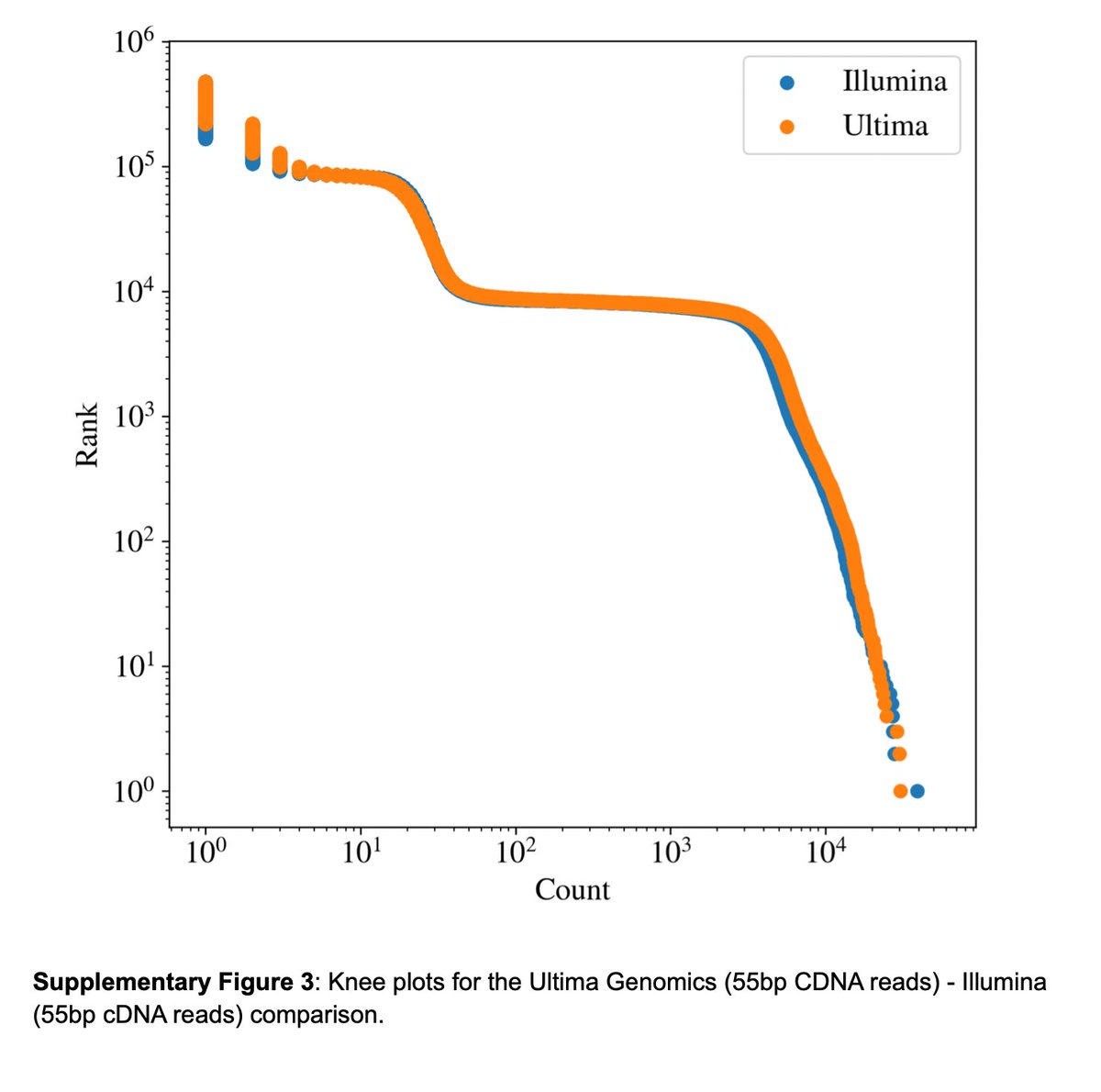
A bit of work but we got an apples-to-apples comparison and to first-order results from the technologies do look similar as claimed. These are the kneeplots: 12/
Next we decided to dig deeper and look at whether there were any genes that differ in the number of counts. There are many. So we picked the top nuclear gene to look at in detail: TMSB4X. It's the 10th most highly expressed gene in the PBMCs assayed. Also an interesting gene. 13/
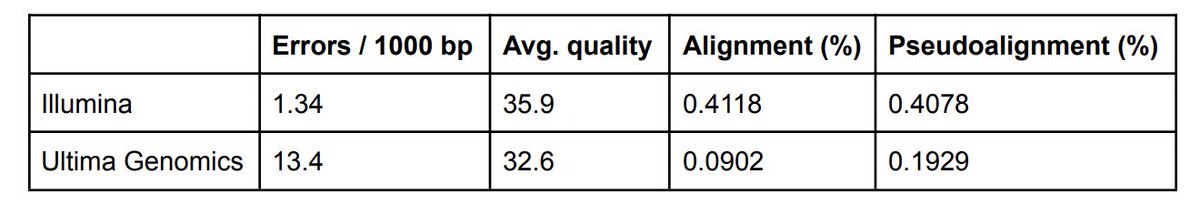
There was a large differential in the number of counts for this gene (how large you might ask... you'll find out in the next tweet!) The differential left us curious as to why. So we took a deep dive into this gene, including aligning (not just pseudoaligning) reads to it. 14/
Results are shown below. The Ultima error rate is 10x Illumina. Thus 4.6x less reads are aligned. As a result, pseudoalignment helps rescue reads (in fact 2.1x more than with alignment), but not all. With Illumina reads there is no diff. between pseudoalignment and alignment. 15/
This gene is worst than most because it has a TTTTTTTT sequence in it (homopolymer of length 8). The technology is terrible at and around such homopolymers (and even shorter ones). An IGV screenshot shows just how bad. 16/
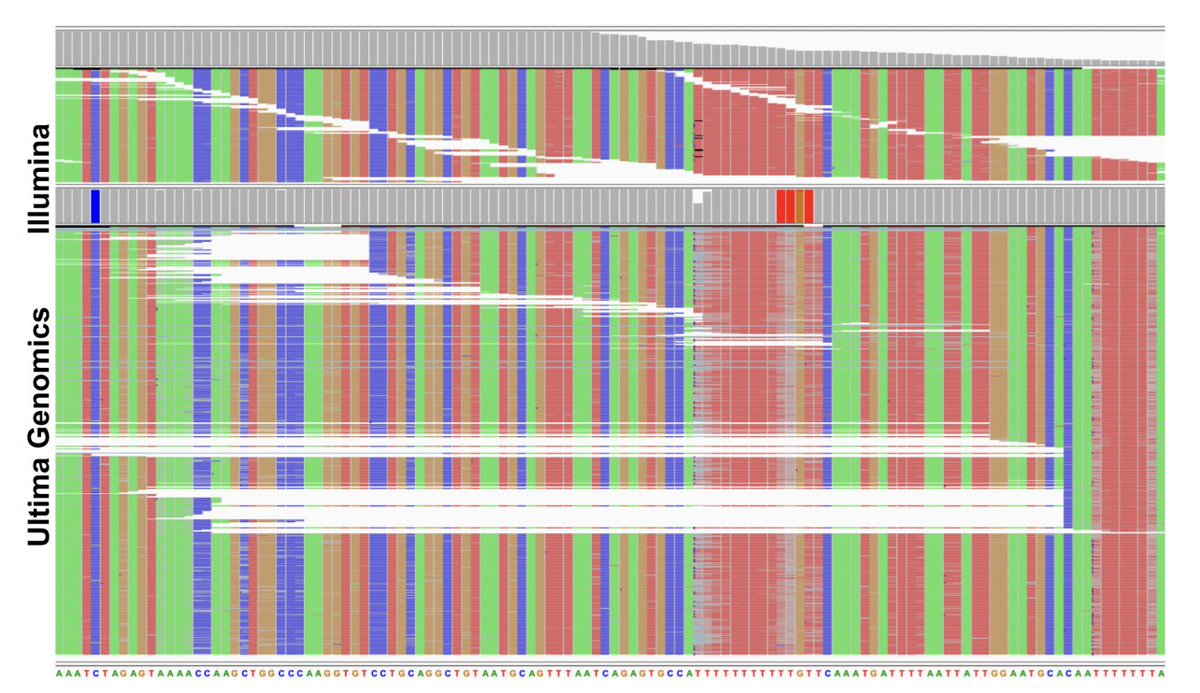
The indel situation particularly bad. 17/
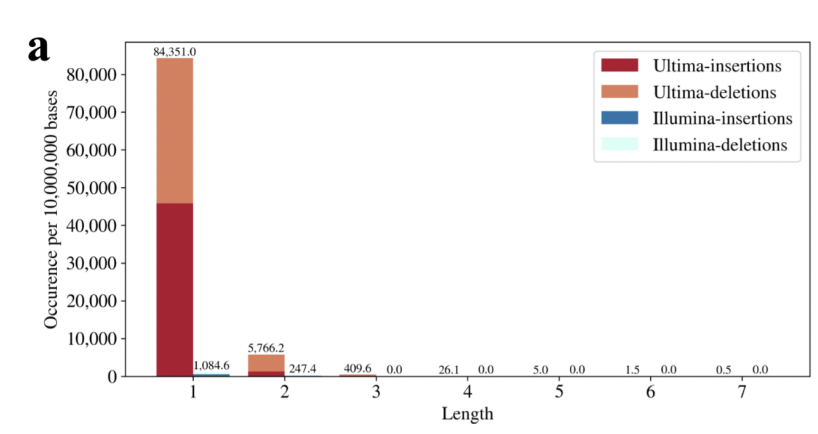
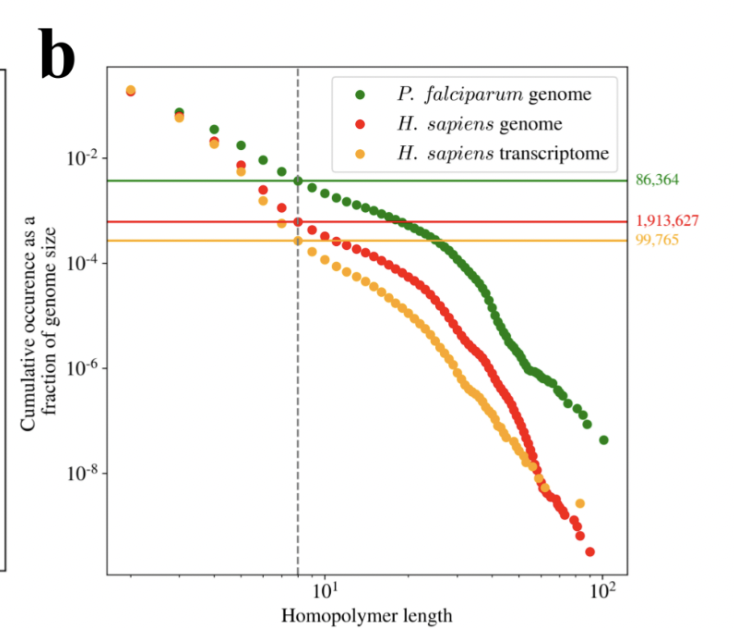
This matters, because with Ultima there is going to be significant bias on certain genes that may be difficult to account and correct for. The human genome is full of homopolymers. We measured how many... lots! 18/
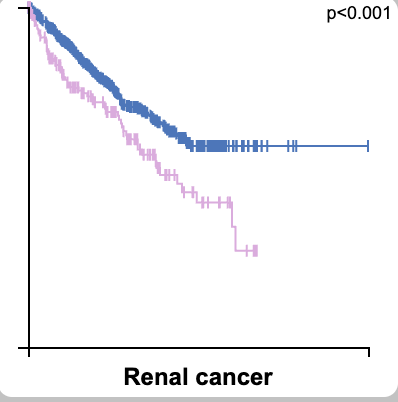
And BTW, TMSB4X may be a renal cancer biomarker, so getting its abundance right matters .❗️ 19/
As for Ultima Genomics, yes, it has a cool new technology that looks faster (which is important), and maybe provides a useful tradeoff of lower cost / higher error rate than Illumina; perhaps useful for assays like #scRNAseq where only read assignment is needed. We'll see. 20/
But the first $100 genome? Ultima Genomics is not first; there is already one available by Nebula. Now I know... this is only 0.4x coverage (30x is $300), but if Ultima Genomics can't sequence homopolymers then is it really $100? 🤷♂️ Also 300/100 = 3 (not 5 or 10). 21/

A few final thoughts: the lack of data release by Ultima Genomics is poor form. It's also disappointing to see many researchers hype the company without looking at the reads. Genome data is still not available. 22/
https://twitter.com/holtjma/status/1531680056707252224
The level of hype is through the roof. @NIH tweeted out that this tech will "ensure that people from ancestrally diverse backgrounds will benefit equitably". Really? Does @NIH think individuals from ancestrally diverse backgrounds have no homopolymers? 23/
https://twitter.com/genome_gov/status/1533827507643985925
Also, what's with the complete omission of any discussion of BGI / MGI? What am I missing? 👀academic.oup.com/nar/article/49… 24/
Tl;dr Ultima Genomics setting up an SBS company is impressive. The speed advance is great. It may be useful for apps where homopolymers don't matter much. And there are interesting compbio challenges ahead to make it better. But error rates are high & the crazy hype is ☹️. 25/25
• • •
Missing some Tweet in this thread? You can try to
force a refresh