🔥Extra-hot pre-print before this hellsite goes up in smoke 🔥
New horizons open up when sequencing costs go ↘️ @UltimaGenomics
Interested in deep cfDNA WGS for ultra-sensitive cancer detection? Duplex sequencing at genome scale?
biorxiv.org/content/10.110…
Check out 🪡👇
New horizons open up when sequencing costs go ↘️ @UltimaGenomics
Interested in deep cfDNA WGS for ultra-sensitive cancer detection? Duplex sequencing at genome scale?
biorxiv.org/content/10.110…
Check out 🪡👇
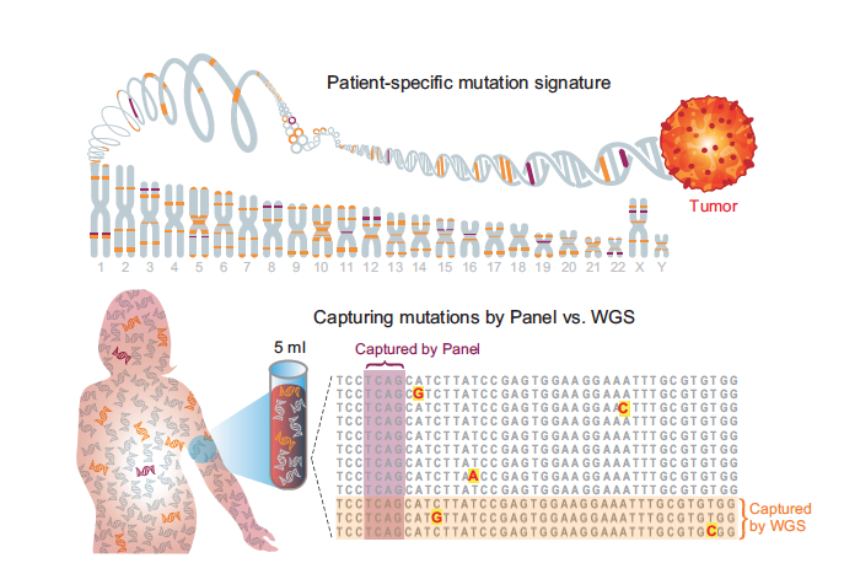
Back in 2020 we introduced the concept of plasma whole genome sequencing (WGS) for MRD detection. We showed that genome wide mutational integration severs the limiting dependency between the number of DNA fragments in the sample and sensitivity.
nature.com/articles/s4159…
nature.com/articles/s4159…

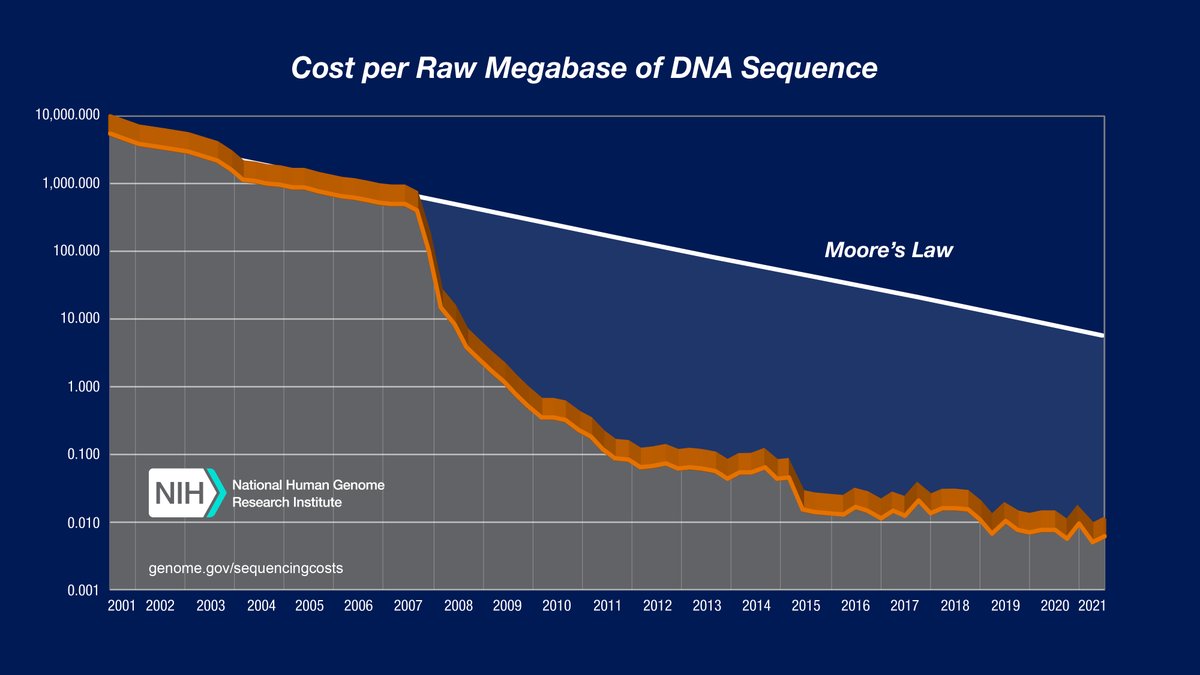
At the time, it was a bit nuts to use WGS bc 💸, but we anticipated that sequencing costs will just keep going down...
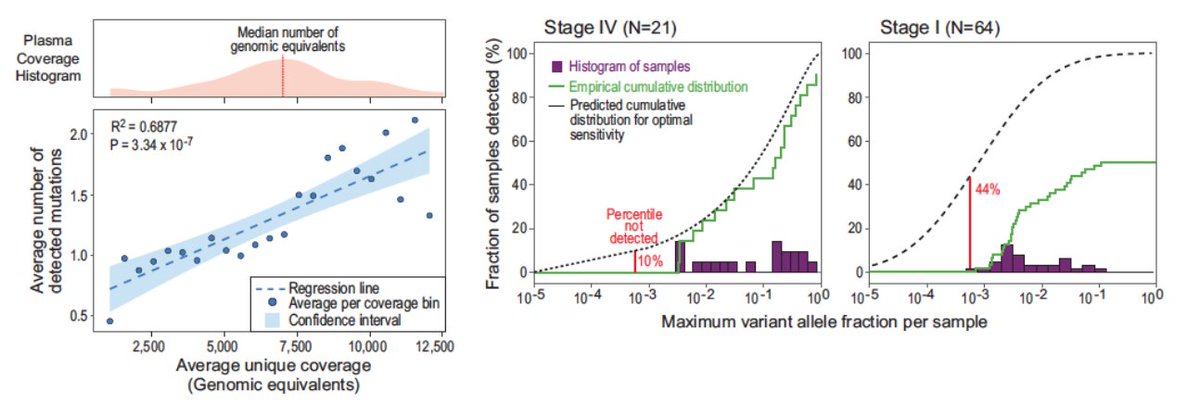
We even made an in silico prediction of how we can have yet greater sensitivity if we can sequence deeper
We even made an in silico prediction of how we can have yet greater sensitivity if we can sequence deeper
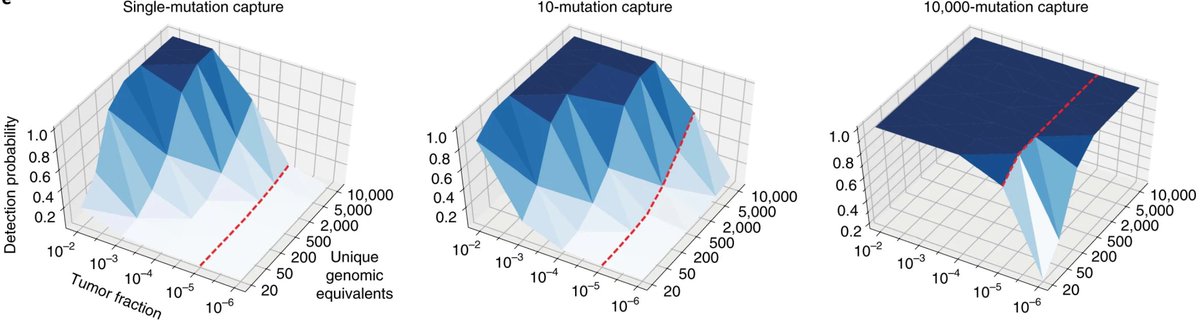
Something changed (let's not go into the why...🫠), and the cost decline stalled...:(
Then we heard 👂about @UltimaGenomics , still deep in stealth mode 🕵️♂️ and got excited...:)
Then we heard 👂about @UltimaGenomics , still deep in stealth mode 🕵️♂️ and got excited...:)
We undertook a comprehensive evaluation comparing short read platforms for plasma WGS and found that @UltimaGenomics delivers clean data for SNV & CNV detection
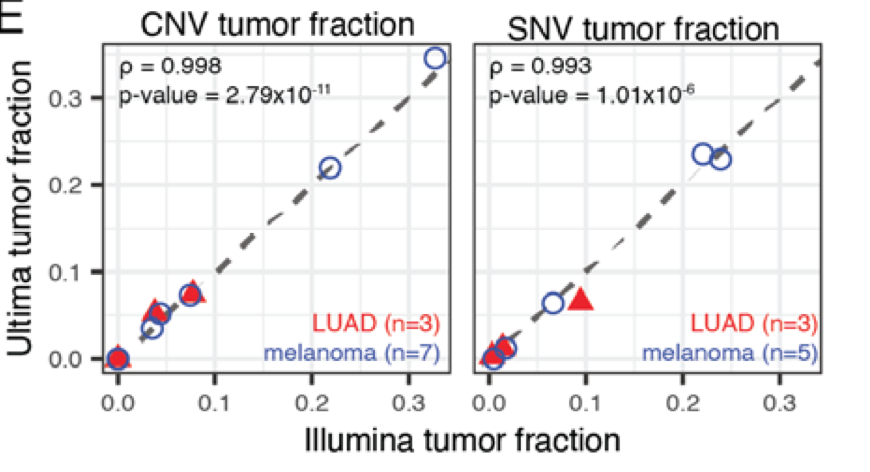
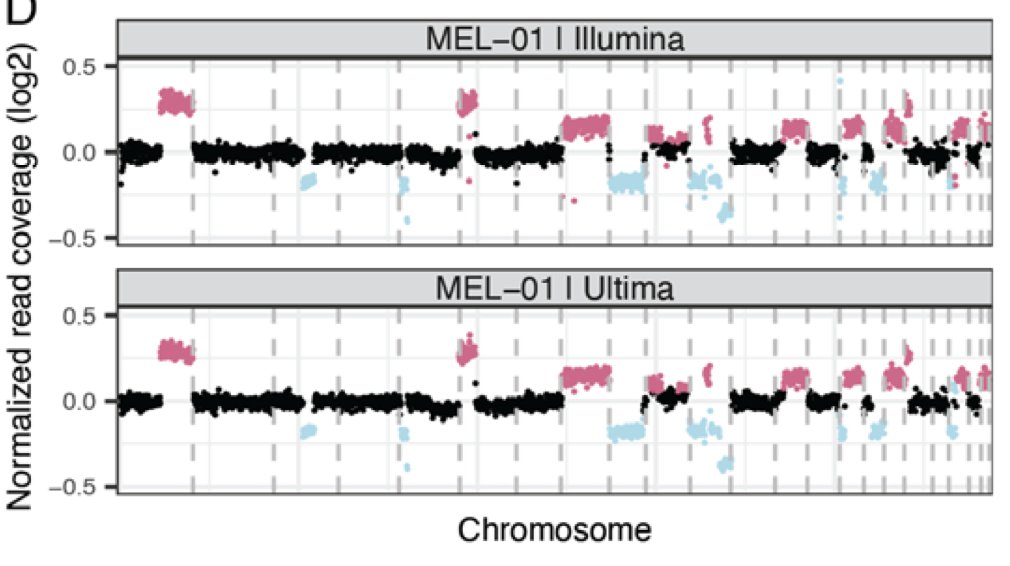
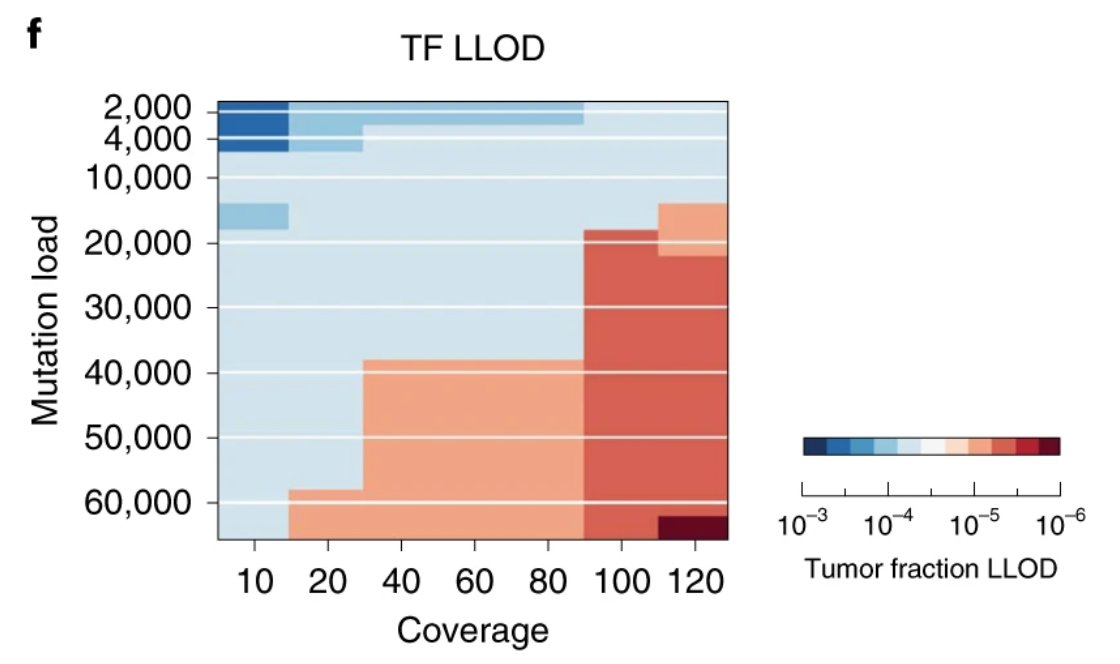
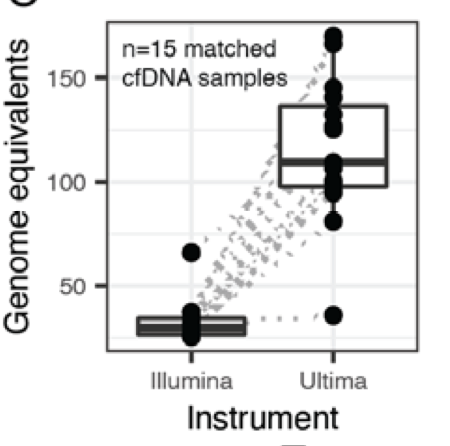
But just as we predicted back then, higher depth allowed us to increase tumor-informed MRD sensitivity to the parts-per-million range!
And it gets wilder...
And it gets wilder...

Duplex error correction has transformed liquid biopsy detection as it allows for single-molecule mutation calling, but is very greedy in sequencing. So we thought, what if we can harness low cost seq. to do plasma Duplex at the scale of the entire genome? 🤯
@AlexandrePCheng and friends figured out how to Duplex on the single-end Ultima reads and showed extra extra clean plasma WGS
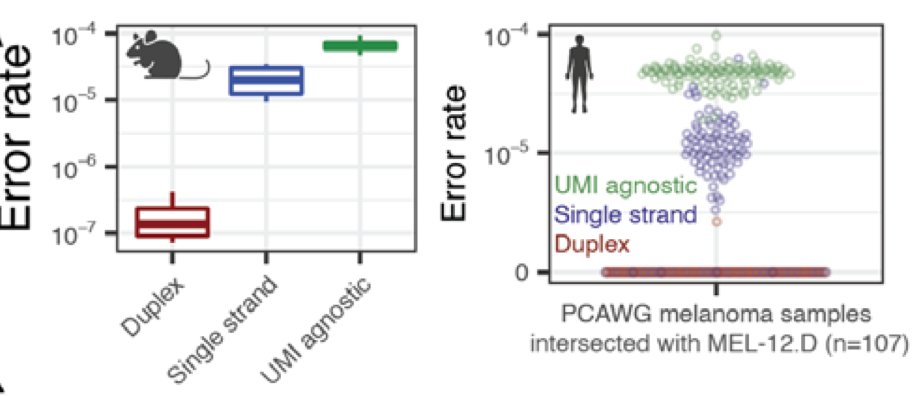
Clean mutational profiles allowed us to match up melanoma plasma samples to a UV signature, and control plasma to a clonal hematopoiesis signature
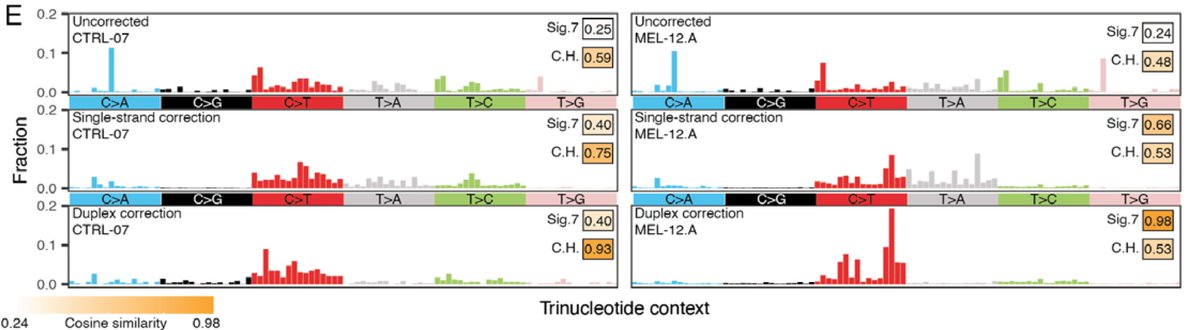
We then leveraged the ability to match genome-wide mutational profiles with mutational signature to compute the relative contribution of ctDNA vs. clonal hematopoiesis, allowing for NON tumor-informed ctDNA detection in the challenging context of low burden melanoma

Super excited about all the possibilities this work opens up in ctDNA and somatic mosaicism. Super excited for the world of declining sequencing costs to be back with us!
@AlexandrePCheng @adamjwidman & A. Arora led this work. Could not have done this without @wolchokj @notSoJunkDNA @gmboland @AltorkiNasser @MPostow @nygenome and many friends. Thank you also to patients and funders!
• • •
Missing some Tweet in this thread? You can try to
force a refresh