
Ran some experiments! #ImmuneBuilder is better than #ESMFold (@alexrives / @TomSercu), right? There are advantages of using the latter, and now a 🧵 on why getting carried away with low RMSDs is only a part of the story...(1/5)
#machinelearning #proteins
#machinelearning #proteins
https://twitter.com/ideasbyjin/status/1593977142748094465
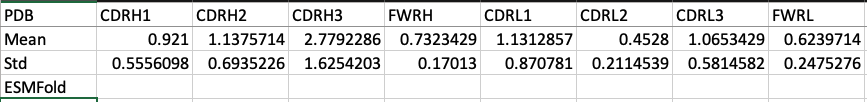

First, the metrics are RMSDs based on aligning the C/N/CA/CB atoms across the chain, then calculating the RMSD across a region. i.e. align every residue of the VH, then calculate RMSD across CDRH3, or CDRH1, etc. This is on ~35 antibodies of the ImmuneBuilder test set (2/5)
ESMFold's CDRH3 accuracies are better than what I expected. Where it's let down is on the "canonical" CDRs. It would've been nice to compare the VH-VL orientations and talk about how ImmuneBuilder doesn't generate D-amino acids, etc. (3/5)
However, antibody-specific tools like ImmuneBuilder tend to -not- model the constant domains, though they are important for binding and function pnas.org/doi/pdf/10.107…; ESMFold can model constant domains, but not the full antibody tail (4/5)
While it's tempting to look at -one- tool as the "top" solution for every problem, again we have to remind ourselves that there are other biologically relevant use cases that only "suboptimal" tools can solve. We really need a biologically-relevant competition soon! (5/5)
• • •
Missing some Tweet in this thread? You can try to
force a refresh




