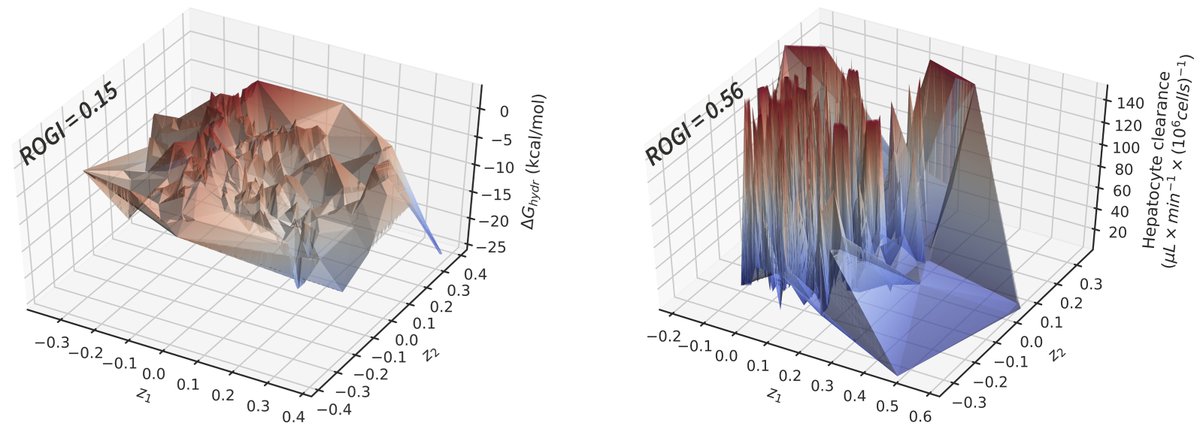
Our team working on ML for chemistry/drug discovery was unfortunately affected by the recent layoffs at Google.
I’m still very much interested in how new technologies can accelerate research in the life and natural sciences.
#ml #ai #compchem #drugdiscovery #googlelayoffs
I’m still very much interested in how new technologies can accelerate research in the life and natural sciences.
#ml #ai #compchem #drugdiscovery #googlelayoffs
If you know of open roles for which expertise in ML, simulation, or modelling as applied to drug/protein/materials design is sought, please feel free to reach out -- any pointer will be much appreciated!
As an opportunity for change, I’m also open to roles that may stretch my skills and expertise. Due to personal constraints, I’ll be looking for positions primarily in the SF Bay and Boston areas.
My colleagues also have considerable experience in ML and drug discovery, and I can’t say enough good things about them. Again, feel free to reach out if aware of potentially suitable positions.
• • •
Missing some Tweet in this thread? You can try to
force a refresh