New preprint on scGPT: first foundation large language model for single cell biology, pretrained on 10 million cells.
Just as text is made of words, cells are characterized by genes
Some thoughts on how cool this is & why it challenges the status quo of single cell analysis🧵🧵
Just as text is made of words, cells are characterized by genes
Some thoughts on how cool this is & why it challenges the status quo of single cell analysis🧵🧵
The self-attention transformer (chatGPT) is so successful in natural language processing (NLP)
But maybe single cell biology is not that different from NLP, as genes & cells correspond to words & sentences?
That's the framing of @BoWang87's awesome paper
biorxiv.org/content/10.110…
But maybe single cell biology is not that different from NLP, as genes & cells correspond to words & sentences?
That's the framing of @BoWang87's awesome paper
biorxiv.org/content/10.110…
Another core similarity between NLP & single cell biology is the large and ever-growing size of publicly available #scRNAseq data (e.g human cell atlas) to be used for training.
Can NLP models also understand intrinsic logics of single cell biology & develop "emergent thinking"?
Can NLP models also understand intrinsic logics of single cell biology & develop "emergent thinking"?
These are compelling parallels, and deep learning tools leveraging them do exist.
For ex, scFormer from @BoWang87 is along similar lines: biorxiv.org/content/10.110…
Another ex. is scBERT: nature.com/articles/s4225…
But AFAIK none of these tools is generative & so versatile as scGPT.
For ex, scFormer from @BoWang87 is along similar lines: biorxiv.org/content/10.110…
Another ex. is scBERT: nature.com/articles/s4225…
But AFAIK none of these tools is generative & so versatile as scGPT.
‼️However: while words in a sentence are sequential, genes in a cell are not.
This is tricky, as GPT predicts the next token auto-regressively, given the sequence of previous tokens.
To understand how this issue is solved here, we'll need to dive into the details of the model.
This is tricky, as GPT predicts the next token auto-regressively, given the sequence of previous tokens.
To understand how this issue is solved here, we'll need to dive into the details of the model.
But before discussing about the methodological details, let's see how scGPT is built and what its performance is on single cell analyses (hint: it does very well).
Different applications correspond each to a different fine-tuning routine, with its own specific objective function
Different applications correspond each to a different fine-tuning routine, with its own specific objective function
During training on 10.3 million scRNAseq blood & bone marrow cells in CellXGene, scGPT simultaneously learns cell & gene representations
The model gradually learns to generate gene expression of cells based on the condition & expression of existing cells.
cellxgene.cziscience.com
The model gradually learns to generate gene expression of cells based on the condition & expression of existing cells.
cellxgene.cziscience.com
The pre-trained model can be fine-tuned to new datasets & specific tasks.
The authors offer fine-tuning pipelines for several tasks commonly done in single cell analysis, s.a. integration, batch correction etc.
Codebase here: github.com/bowang-lab/scG…
The authors offer fine-tuning pipelines for several tasks commonly done in single cell analysis, s.a. integration, batch correction etc.
Codebase here: github.com/bowang-lab/scG…

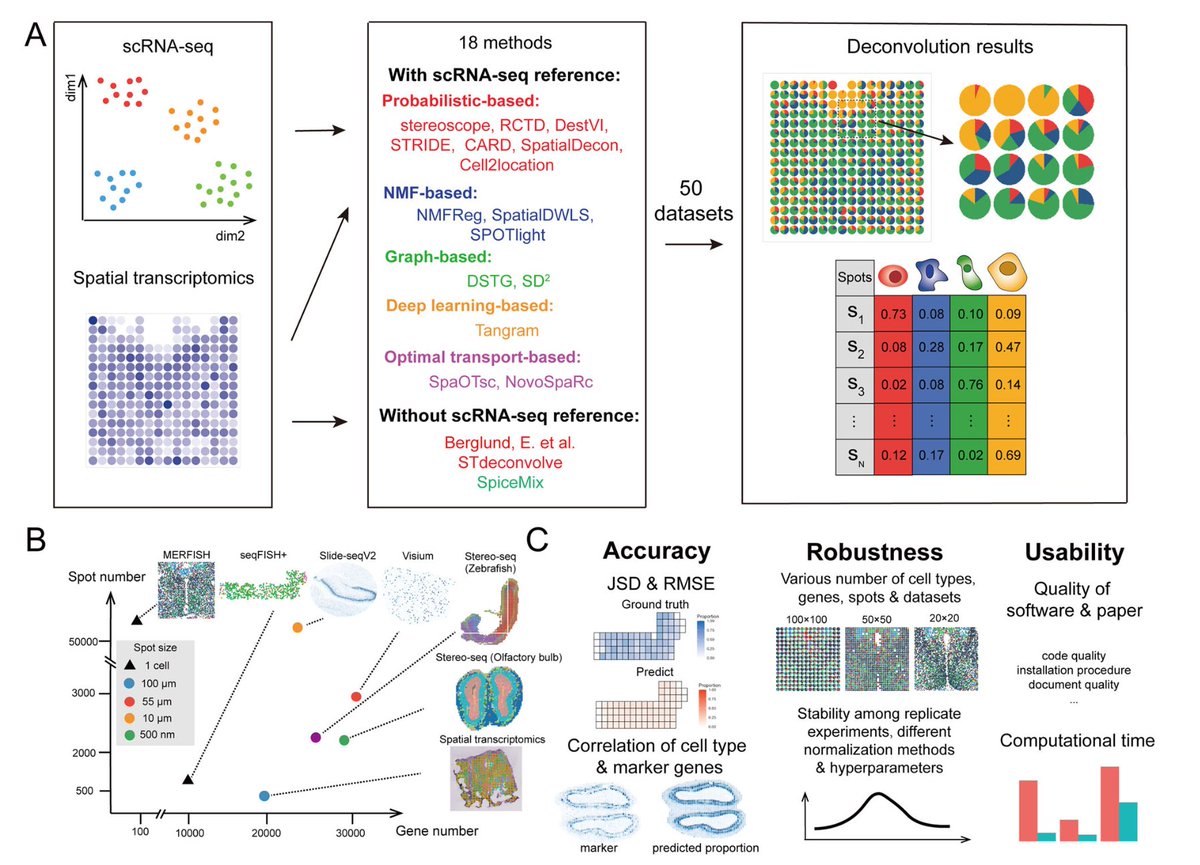
The single cell genomics tasks evaluated in the preprint are:
1. integration & batch correction
2. cell type annotation
3. genetic perturbation prediction
4. multi-omic integration
5. gene regulatory networks inference
1. integration & batch correction
2. cell type annotation
3. genetic perturbation prediction
4. multi-omic integration
5. gene regulatory networks inference
1. Integration of multiple scRNA-seq datasets with batch correction
scGPT was benchmarked against scVI (also a deep learning model), Harmony and Seurat, on integrating two datasets: PBMC (2 batches) & Immune Human (10 batches).
scGPT was benchmarked against scVI (also a deep learning model), Harmony and Seurat, on integrating two datasets: PBMC (2 batches) & Immune Human (10 batches).

scGPT performed best, as assessed by multiple biological conservation metrics (remember, the goal here is to minimize the spread of cells of same cell type).
scGPT (dark pink) is consistently a higher bar than the rest.
Still, all tools seem to do pretty well generally❗
scGPT (dark pink) is consistently a higher bar than the rest.
Still, all tools seem to do pretty well generally❗

2. Cell type annotation
For this task, the pre-trained scGPT model was fine-tuned using cross-entropy loss against ground-truth labels from a new reference dataset of human pancreas cells.
It was then tasked to identify cell types on another human pancreas dataset.
For this task, the pre-trained scGPT model was fine-tuned using cross-entropy loss against ground-truth labels from a new reference dataset of human pancreas cells.
It was then tasked to identify cell types on another human pancreas dataset.
scGPT did very well on this task, classifying cells almost perfectly.
However, the cell type labels to be inferred are pretty general & biologically distinct, so separating them is quite easy, regardless of how strong the model is.
However, the cell type labels to be inferred are pretty general & biologically distinct, so separating them is quite easy, regardless of how strong the model is.

Interesting to now pause & reflect for a bit.
We've just seen that also chatGPT can do cell type assignment, by essentially literature browsing
However, there's a very important difference here
scGPT is, in a sense, the opposite of literature browsing, as it is fully automatic
We've just seen that also chatGPT can do cell type assignment, by essentially literature browsing
However, there's a very important difference here
scGPT is, in a sense, the opposite of literature browsing, as it is fully automatic
https://twitter.com/simocristea/status/1651637310012936193
3. Perturbation prediction
Two Perturb-Seq datasets, on which the correlation between predicted & corresponding ground-truth expression values after perturbation was evaluated.
scGPT did well, but (again) all tested tools did have similar performance❗
Two Perturb-Seq datasets, on which the correlation between predicted & corresponding ground-truth expression values after perturbation was evaluated.
scGPT did well, but (again) all tested tools did have similar performance❗

4. Multi-omic integration
Each omic type (e.g gene expression, chromatin accessibility, protein abundance) corresponds to a different language in NLP.
scGPT did very well (by multiple biological arguments), e.g it is the only method to produce a separate cluster for CD8 naive T
Each omic type (e.g gene expression, chromatin accessibility, protein abundance) corresponds to a different language in NLP.
scGPT did very well (by multiple biological arguments), e.g it is the only method to produce a separate cluster for CD8 naive T

5. Gene Regulatory Network inference
On this task, scGPT was tested to group functionally related genes & differentiate distinct genes from its gene embedding network.
The preprint also shows improvement of fine-tuning on the Immune Human dataset vs. pre-trained only.
On this task, scGPT was tested to group functionally related genes & differentiate distinct genes from its gene embedding network.
The preprint also shows improvement of fine-tuning on the Immune Human dataset vs. pre-trained only.
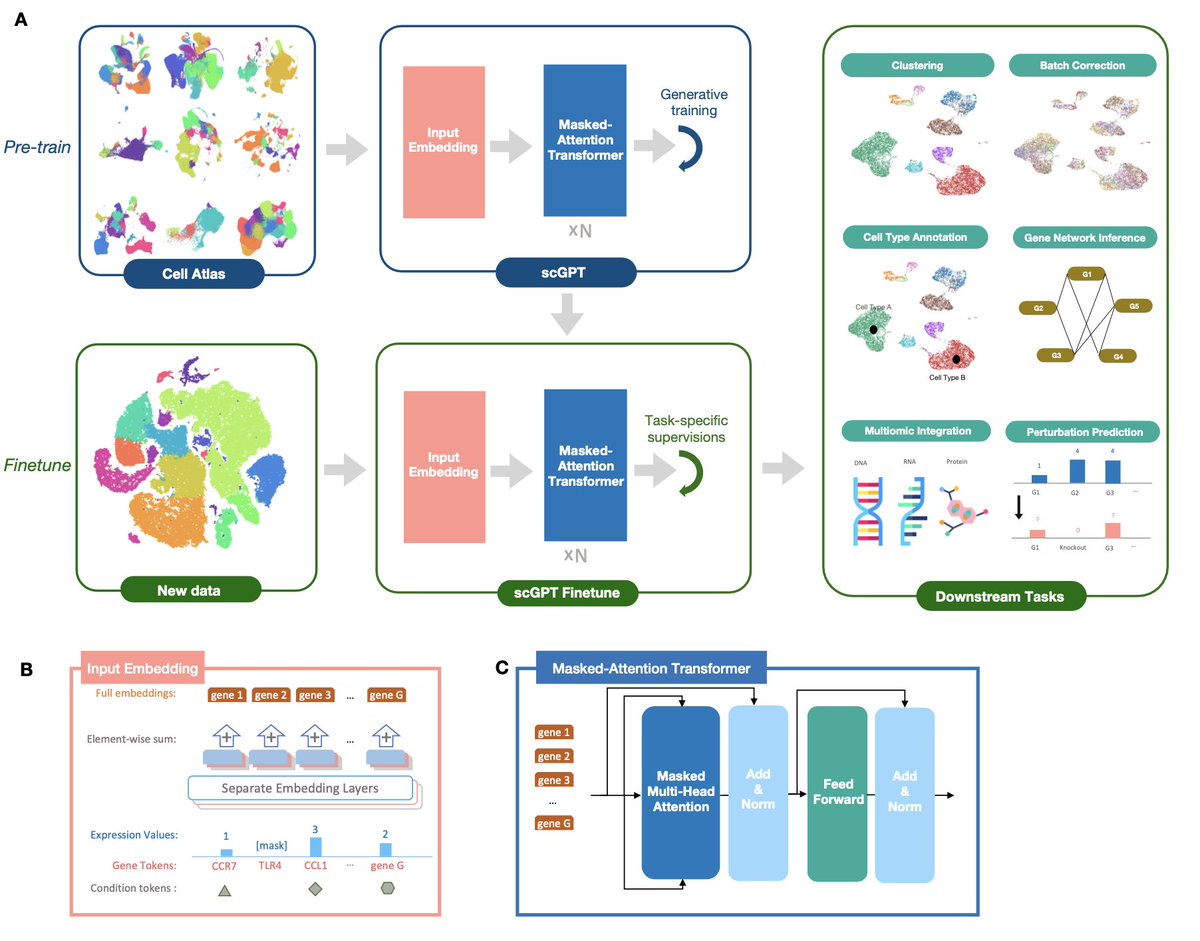
Now that we’ve seen scGPT has great performances, let's discuss some details about the model.
First, its 3 inputs:
1. gene tokens
2. expression values
3. condition tokens
Genes are the smallest unit of information, equivalent to a word in natural language generation.
First, its 3 inputs:
1. gene tokens
2. expression values
3. condition tokens
Genes are the smallest unit of information, equivalent to a word in natural language generation.

The gene names are used as tokens.
Each gene is assigned a unique integer identifier id(gj) within the complete vocabulary of tokens.
The input gene tokens of each cell i are a vector tg(i) of pre-defined length M = the (variable) number of highly variable genes.
Each gene is assigned a unique integer identifier id(gj) within the complete vocabulary of tokens.
The input gene tokens of each cell i are a vector tg(i) of pre-defined length M = the (variable) number of highly variable genes.

This conceptual parallel is nice & easy, as different sets of gene tokens can be integrated into a common vocabulary by taking the union set of all genes across all the studies analyzed in an application (with however potential computing restrictions on M during pre-training).
Next, the authors choose to normalize their input data (raw counts) in a different manner than the usual normalization routines (e.g TPM or log1p).
The argument is that absolute values can convey different "semantic" meanings in different scenarios (for example multiple batches)
The argument is that absolute values can convey different "semantic" meanings in different scenarios (for example multiple batches)
This approach is both interesting & quite extreme: the raw counts for each cell are separated into a number B of bins according to expression values
All genes within a bin get assigned the same value, which is the index of the bin( e.g all genes in bin number 10 are assigned 10)
All genes within a bin get assigned the same value, which is the index of the bin( e.g all genes in bin number 10 are assigned 10)
Before binning, the log1p transformation is applied, followed by selecting M most highly variable genes.
Therefore: the input for cell i are log1p expression values of the M highest variable genes, binned across B bins (not sure about actual values of B).
Here's what this means
Therefore: the input for cell i are log1p expression values of the M highest variable genes, binned across B bins (not sure about actual values of B).
Here's what this means

Binning the trades noise reduction with removing biological variability.
I am curious how impactful this step is on the model's performance & how other normalizations do.
Tuning the normalization might be a way to improve the model.
@BoWang87 any thoughts?
I am curious how impactful this step is on the model's performance & how other normalizations do.
Tuning the normalization might be a way to improve the model.
@BoWang87 any thoughts?
Another interesting strategy is that, during pre-training, the input is restricted to only genes with non-zero expression for each input cell.
On the contrary, during fine-tuning, all genes (both zero and non-zero) are included.
On the contrary, during fine-tuning, all genes (both zero and non-zero) are included.
The model also accommodates gene condition tokens, which are indicators of class belonging among the genes, such as belonging to functional pathways or perturbation alterations.
Each cell is considered a "sentence" composed of genes, and its representation is obtained by aggregating the gene-level representations.
As when representing an NLP transformer, there is a special token that indicates cell membership, i.e. which genes belong together to a cell.
As when representing an NLP transformer, there is a special token that indicates cell membership, i.e. which genes belong together to a cell.
The model allows adding condition tokens among cells as well, for indicating different sequencing modalities, batches, perturbation states & others.
The way this is modeled is as a gene-level token, repeated M times for each cell i.
The way this is modeled is as a gene-level token, repeated M times for each cell i.
The cell-level tokens are not used as input to the transformer blocks, rather concatenated with the transformer output before fine-tuning (e.g. concatenation of cell representation with batch embedding in the task of scRNAseq integration).
Finally, how about the sequentiality among genes, as for text generation?
The answer here is a specialized attention masking procedure that defines the *order of prediction* from attention scores.
This is an innovative idea, which also helps capture interactions among genes.
The answer here is a specialized attention masking procedure that defines the *order of prediction* from attention scores.
This is an innovative idea, which also helps capture interactions among genes.
It works by iteratively predicting expression of a new genes set, which in turn become the “known genes” in next iteration of attention computation.
In iterative generation rounds, this creates an order among genes from prediction confidence, mimicking auto-regressive generation
In iterative generation rounds, this creates an order among genes from prediction confidence, mimicking auto-regressive generation
That's what scGPT is all about!
Now, clear future directions:
1. Multiple normal cell types (not only immune) - Likely easy
2. Disease - Likely very difficult, but also very interesting (think cancer heterogeneity)
3. Spatial location - can it also be learned & generated?
Now, clear future directions:
1. Multiple normal cell types (not only immune) - Likely easy
2. Disease - Likely very difficult, but also very interesting (think cancer heterogeneity)
3. Spatial location - can it also be learned & generated?
The single cell genomics community has meany reasons to be hyped. Not only about this pre-print itself, but also about what it represents, and especially the potential of such type of models.
There’s many things that we have no idea how to understand or predict in biology.
LLMs are a new shot at this complex problem.
Intuitively, it’s a very exciting direction to discover.
LLMs are a new shot at this complex problem.
Intuitively, it’s a very exciting direction to discover.
It’s impressive that scGPT performs so well on many different tasks. Still, its performance is comparable (indeed better!) with other existing tools.
So it’s natural to ask oursevles: is there any real gain? Are we willing to trade interpretability for modest performance gains?
So it’s natural to ask oursevles: is there any real gain? Are we willing to trade interpretability for modest performance gains?
In fact, the biggest advantages of this framework are its modularity & scalability.
On the basis of a single pre-trained model, multiple different pre-training routines, corresponding to different single cell applications, can be built relatively easy.
This is nicely extendable
On the basis of a single pre-trained model, multiple different pre-training routines, corresponding to different single cell applications, can be built relatively easy.
This is nicely extendable
There are no words on how excited I am about the potential of applying this framework to disease tissues & understanding the complex language of cancer genomics.
This preprint was a great read, looking forward for trying out the finetuned models!
Congrats 👏 @BoWang87 & team!
This preprint was a great read, looking forward for trying out the finetuned models!
Congrats 👏 @BoWang87 & team!
• • •
Missing some Tweet in this thread? You can try to
force a refresh