PhD. Systems Bio, comparing things to each other, protein language models, plants, evo, proteomics. Princeton, Lewis-Sigler Scholar, prev: UT Austin
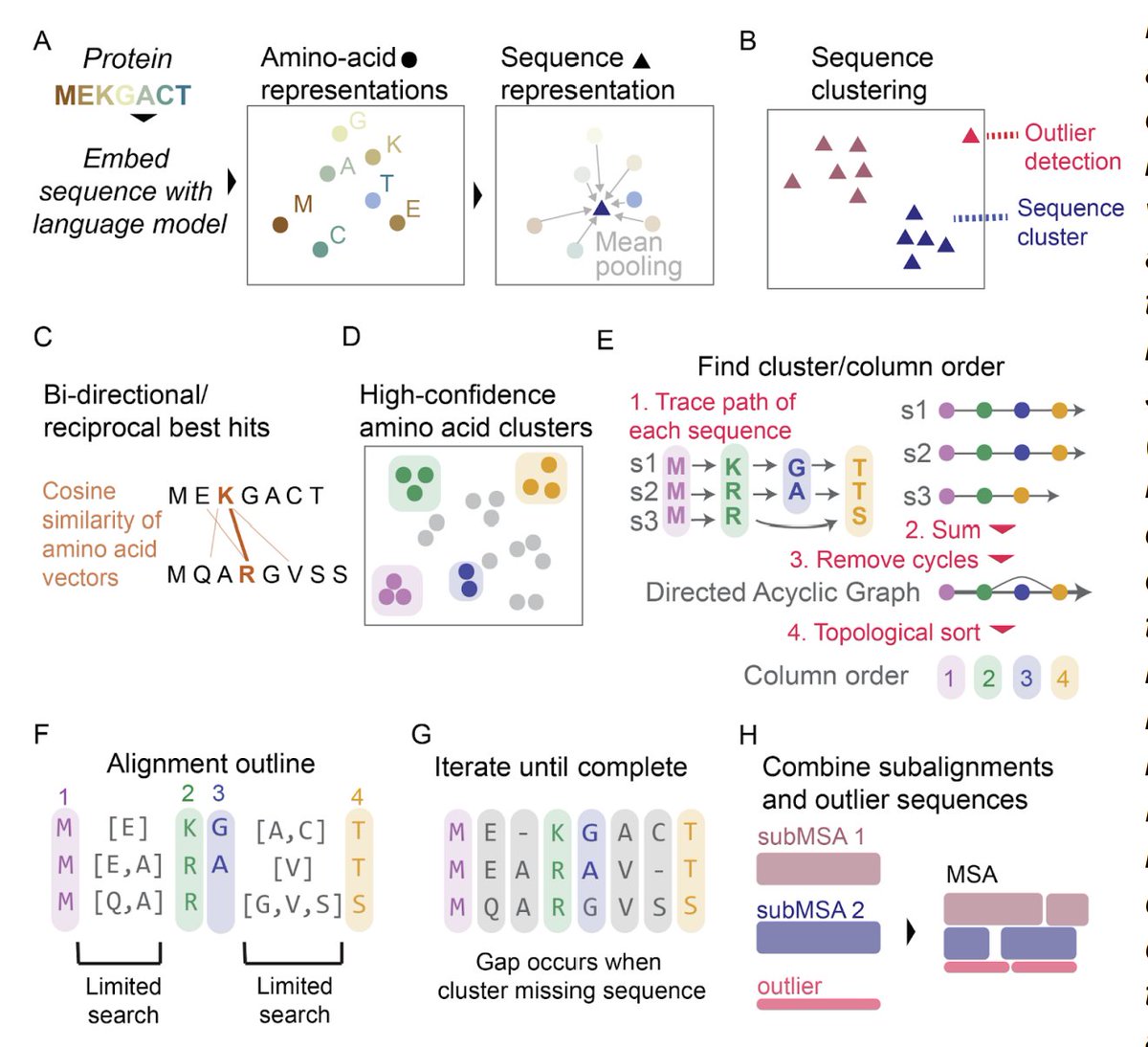 Language models output a long numeric vector for each amino acid in a sequence. Amino acids at equivalent positions in different sequences should have similar vectors. Clustering these vectors is the basis of vcMSA (vector-clustering Multiple Sequence Alignment).
Language models output a long numeric vector for each amino acid in a sequence. Amino acids at equivalent positions in different sequences should have similar vectors. Clustering these vectors is the basis of vcMSA (vector-clustering Multiple Sequence Alignment). 

 First approach was putting the unclustered graph into Large Graph Layout. I have to consult my own blogpost to run the program, so I guess write for yourself clairemcwhite.github.io/lgl-guide/. The graph is too dense for even the magic bullet of LGL to get good separation w/o clustering 2/5
First approach was putting the unclustered graph into Large Graph Layout. I have to consult my own blogpost to run the program, so I guess write for yourself clairemcwhite.github.io/lgl-guide/. The graph is too dense for even the magic bullet of LGL to get good separation w/o clustering 2/5