
An update on #SARSCoV2 selection analysis using @GISAID data (observablehq.com/@spond/natural…). I added a simple 5-category classification for each potential interesting site. One category = one point. The more points, the more interesting a site is.
Category 1. Is the site under selection using statistical comparative methods?
Category 2. Is there a large (>20%, which is incidentally what you can detect with mixed bases) fraction of minority alleles (synonymous or non-synonymous) among viral haplotypes at the site.
Category 2. Is there a large (>20%, which is incidentally what you can detect with mixed bases) fraction of minority alleles (synonymous or non-synonymous) among viral haplotypes at the site.
Category 3. Is there an upward trend over time in how many sequences carry a variant, i.e. do we see that variant frequency is increasing over time?
Category 4. Do we see multiple evolutionary events on the tree, i.e. more than one internal branch with selection?
Category 4. Do we see multiple evolutionary events on the tree, i.e. more than one internal branch with selection?
Category 5. Is there any evidence, either from NGS data (covid19.galaxyproject.org/genomics/4-Var…) or mixed bases in genomic sequences (e.g. CYT for CCT or CTT, P/L) to suggest that within-host the same position is variable? Adaptation starts within host, so this is a critical piece
And, curiously, @art_poon @sdwfrost and I had a paper on using mixed bases to look for intra-host/inter-host adaptation (journals.plos.org/plospathogens/…). I am disappointed that we are back to using Sanger-level resolution from NGS data.
Every daily analysis will now sort positions based on their cumulative category score (each residue listed must at a minimum be either statistically selected or have high MAF - minor allele frequency) 

There are two 5-star sites, nsp6 37 and RdRp 323, which are selected, have high MAF, increasing frequencies over time, multiple evolutionary events on the tree, and NGS intra-host data. Interestingly, a top 5 site is nsp3 106 which is negatively selected.
You can also use the notebook now to view the geographic distribution of each variant residue, and the time trends for said residue. For example, here's the overall distribution of the ancestral residue P at RdRp 323

And the "derived" residue L

L over data from March 13 or later
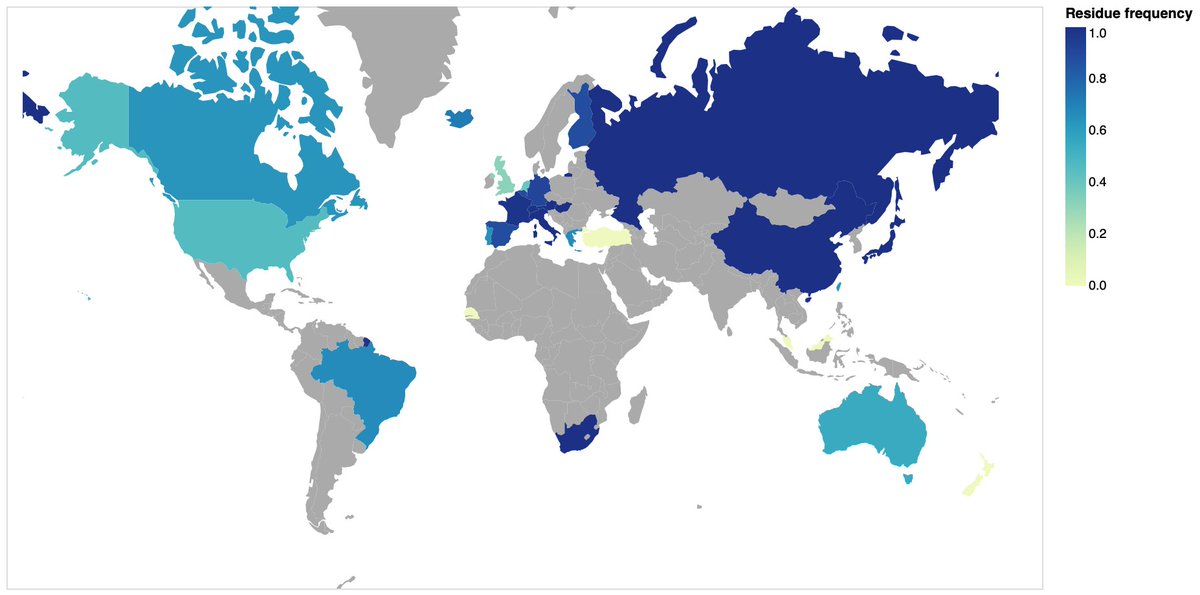
You can also see which countries have genomes with mixed bases (P or L in this case)

This week, I'll be adding another category: evidence of selection using comparisons with related coronavirus sequences.

