Dad. Husband. Born/raised Kyiv. Developer of @hyphy_software. Former mathematician, current professor of biology at Temple U. Google scholar: https://t.co/qyzpbkE1qZ




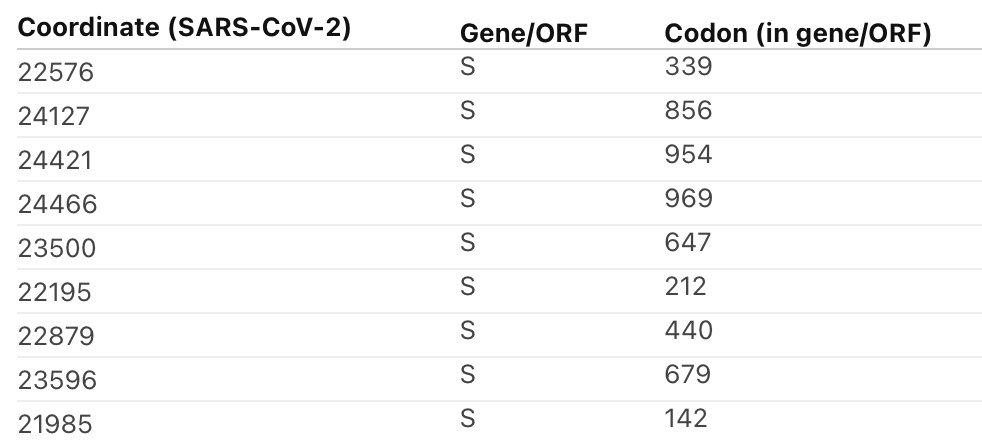
 2/6 Selection pressure on #Omicron seems to have *stabilized* both in terms which sites are under selection and the overall dN/dS estimates, where dN/dS on internal branches is dropping down where the leaves are (dN/dS internals > dN/dS leaves was very unusual)
2/6 Selection pressure on #Omicron seems to have *stabilized* both in terms which sites are under selection and the overall dN/dS estimates, where dN/dS on internal branches is dropping down where the leaves are (dN/dS internals > dN/dS leaves was very unusual) 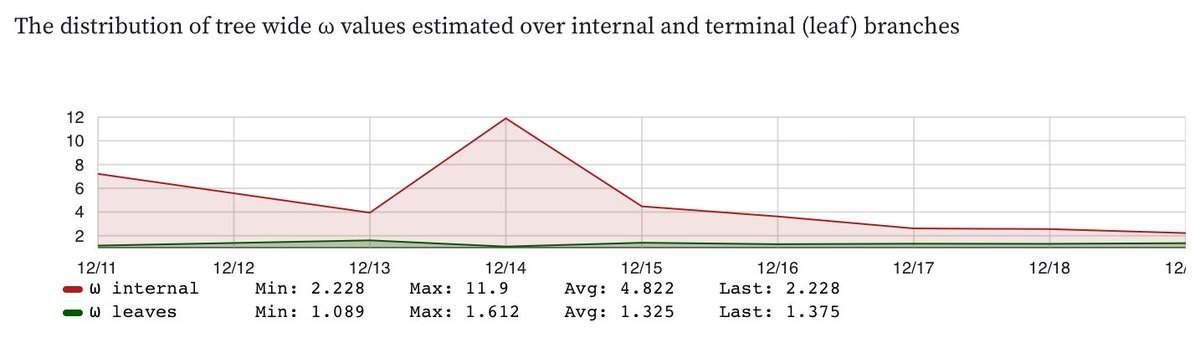
 2/4 S/1081 is perfectly conserved (I) in sarbecoviruses (@SpyrosLytras), and "V" was found in ~100 isolates prior to #Omicron. It is at ~5% in BA.1 sequences.
2/4 S/1081 is perfectly conserved (I) in sarbecoviruses (@SpyrosLytras), and "V" was found in ~100 isolates prior to #Omicron. It is at ~5% in BA.1 sequences.



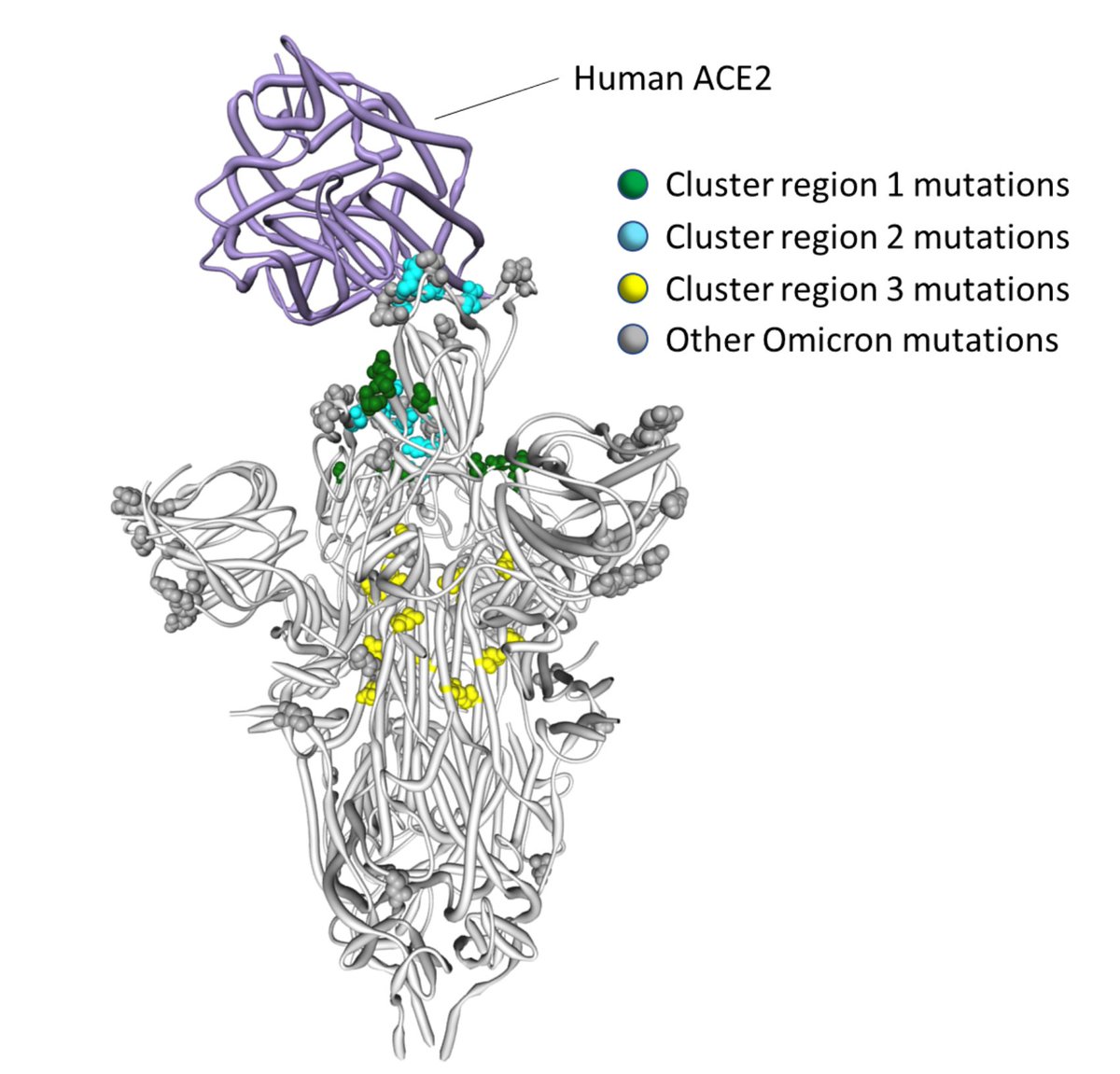
 2/4 In particular, S/796 has experienced what we termed "stepwise evolution" in SARS-CoV-1 and is near the trimerization surface, which undergoes conformational rearrangements during viral fusion
2/4 In particular, S/796 has experienced what we termed "stepwise evolution" in SARS-CoV-1 and is near the trimerization surface, which undergoes conformational rearrangements during viral fusion