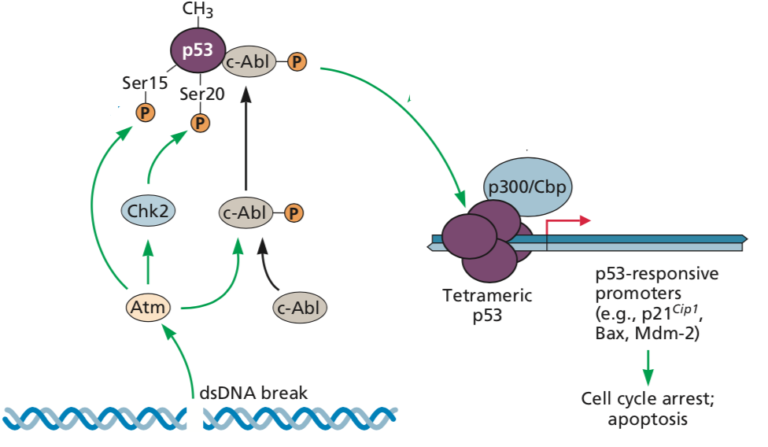
Very excited to share our synthetic lethality based precision oncology approach via the tumor transcriptome (SELECT) @Cell
This effort is led by @joo_sang_lee @theNCI #PrecisionMedicine [1/n]
cell.com/cell/fulltext/…
This effort is led by @joo_sang_lee @theNCI #PrecisionMedicine [1/n]
cell.com/cell/fulltext/…
Current precision oncology mainly targets actionable mutations & has limited patient coverage. We present the first precision oncology framework that systematically guides cancer treatment based on *synthetic lethal* vulnerabilities identified via the tumor transcriptome.
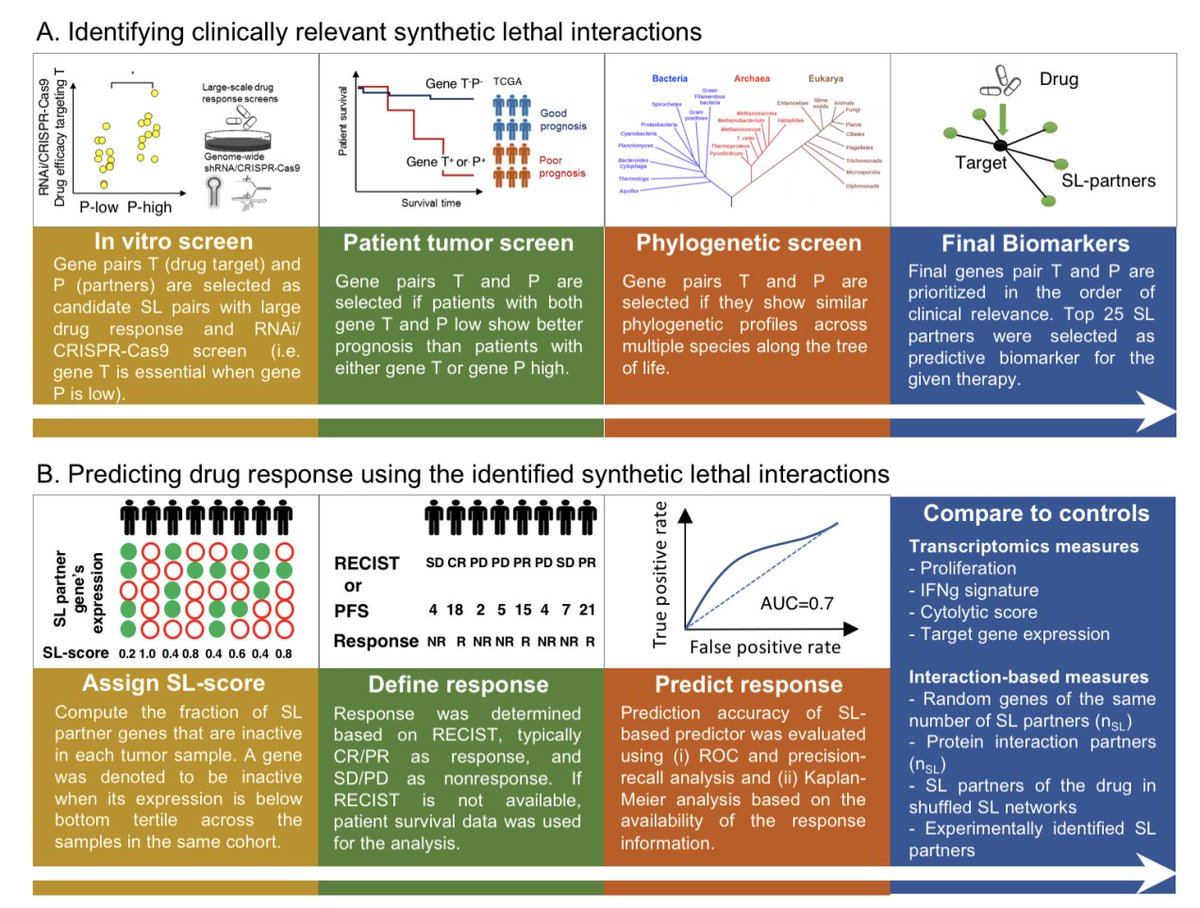
*Synthetic lethality* (SL) is an interaction between a pair of genes, where their individual inactivation has no phenotype, but their co-inactivation leads to cell death. We identified the SL pairs from analyzing a large collection of patient cohorts of 10k tumors. 

The basic idea underlying the use of SLs for guiding patients is that a drug is likely to be more effective in patients where many of the SL partners of its targets are under-expressed in the tumor, thus leading to specific and selective tumor cell death.

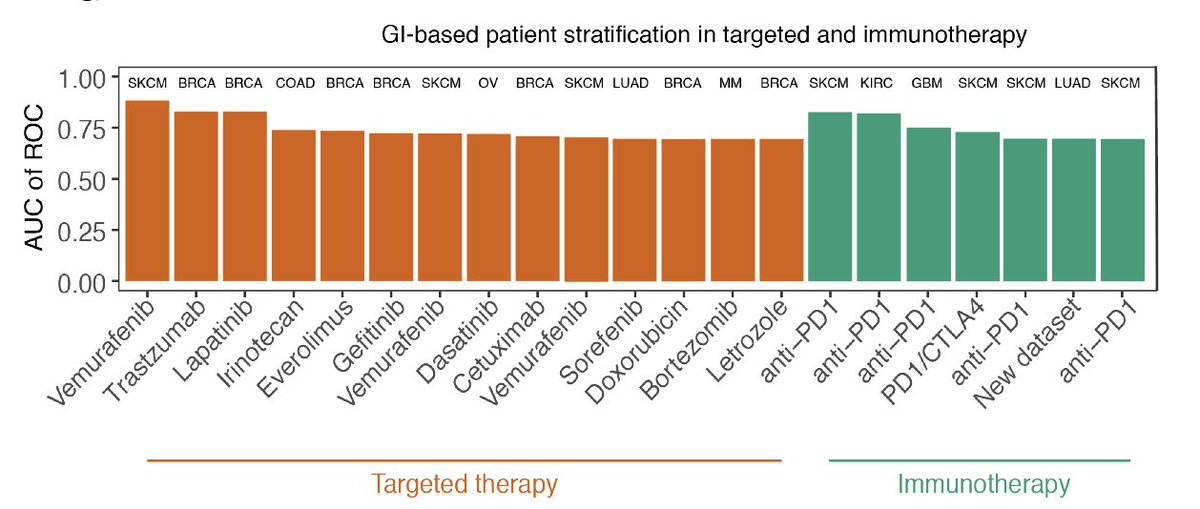
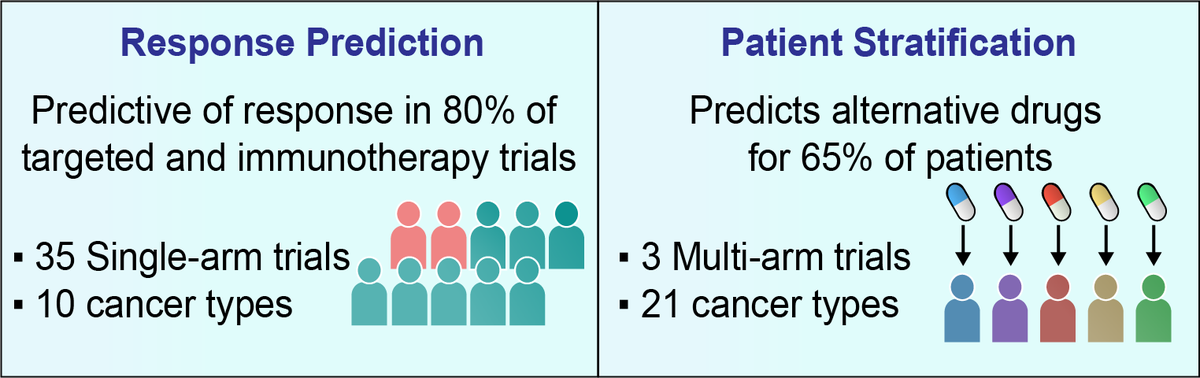
SELECT prediction accuracy was tested on a large collection of 35 clinical trial datasets (spanning 14 targeted therapy and 21 immunotherapy cohorts across 10 cancer types), a scale markedly surpassing previous efforts to our knowledge.
For targeted therapy, SELECT is predictive of patients’ tumor response in 7 out of 10 clinical trial datasets with accuracy levels of AUC of ROC>0.7 and successfully predicts patient survival in 3 out of 4 additional cohorts.

For checkpoint immunotherapy, SELECT is predictive of tumor response in 15 out of 18 clinical trial datasets with accuracy levels of AUC>0.7 and successfully predicts patient survival in 3 additional cohorts (Gide et al. also shows AUC>0.7 in addition to the survival signal).

This collection includes a fresh pre-treatment transcriptomics profiles and treatment response data from Samsung Medical Center, where lung adenocarcinoma patients were treated with pembrolizumab (ncbi.nlm.nih.gov/geo/query/acc.…).
Overall, through a retrospective analysis of 35 targeted or immunotherapy clinical trials of about 2,500 patients, SELECT is predictive of response in 28 out of 35 cases (80%) across 10 different cancer types.

Evaluating this approach retrospectively in a recent WINTHER multi-arm basket clinical trial, we show that SELECT is predictive of the response and that the fraction of patients benefitting from treatments may possibly be markedly increased by targeting SL vulnerabilities.

The attached figure illustrates reports that a clinician would receive from a SELECT analysis. Based on pre-treatment transcriptomic profiles of patients, an SL-score will be assigned to each drug, providing clinicians with personalized SL-based treatment options.

SELECT is the first synthetic lethality-based precision oncology tool for analyzing the tumor transcriptome. It shows considerable accuracy across many different targeted and immunotherapy trials, laying a basis for future prospective transcriptomics-based clinical trials.
Without the dedication of clinicians and patients involved in all the trials analyzed in this study, this research wouldn’t have been possible. We greatly thank them and made our codes and datasets publicly available for academic purposes (zenodo.org/record/4661265).
Many thanks to our co-authors @NishanthUNair @KunWang331 @Sanjusinha7 and others, and thanks to our wonderful collaborators @NCIKenAldape @Dr_R_Kurzrock @MRGneuroonc @hannenhalli @ZeevRonai and many more!! Read the full paper @CellCellPress :)
• • •
Missing some Tweet in this thread? You can try to
force a refresh