Here is our assessment of #SARSCoV2 #Omicron receptor usage and immune evasion in collaboration with the team of @DavideCorti6 @Vir_Biotech
Led by Elisabetta Cameroni, Christian Saliba, @johnbowenbio & Laura E. Rosen
@HHMINEWS @UWBiochemistry
biorxiv.org/content/10.110…
1/8
Led by Elisabetta Cameroni, Christian Saliba, @johnbowenbio & Laura E. Rosen
@HHMINEWS @UWBiochemistry
biorxiv.org/content/10.110…
1/8
#SARSCoV2 #Omicron harbors a staggering 37 amino acid mutations in the spike with 15 of them in the receptor-binding domain (RBD), which is the main target of neutralizing antibodies. The number and positions of these mutations is concerning for tropism & immune evasion.
2/8
2/8
We found that the #SARSCoV2 #Omicron RBD has ~2.5-fold enhanced ACE2 binding affinity, relative to the Wuhan-Hu-1 RBD, similar to what we previously showed for the Beta variant of concern.
nature.com/articles/s4158…
3/8
nature.com/articles/s4158…
3/8

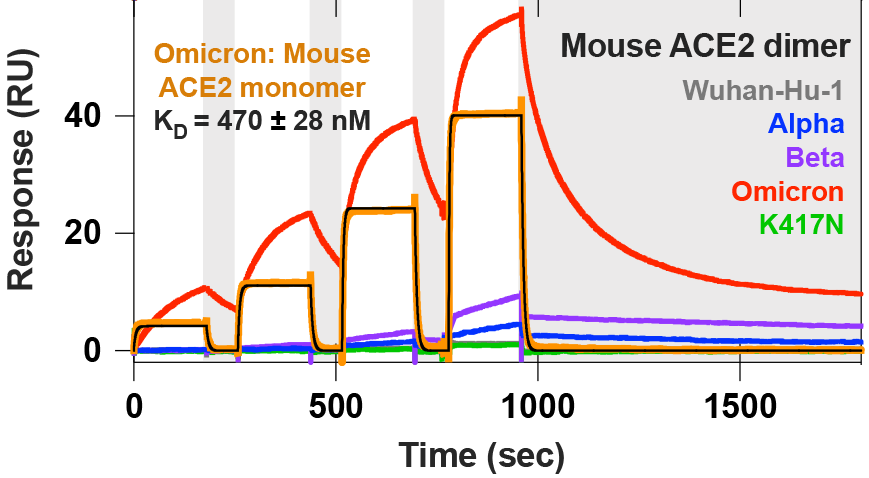
We discovered that the #SARSCoV2 #Omicron RBD, but not Alpha or Beta RBDs, bound mouse ACE2, likely due to Q493R which is similar to Q493K isolated upon mouse adaptation by @Baric_Lab
sciencedirect.com/science/articl…
These data *putatively* reflect #Omicron expanded tropism
4/8
sciencedirect.com/science/articl…
These data *putatively* reflect #Omicron expanded tropism
4/8
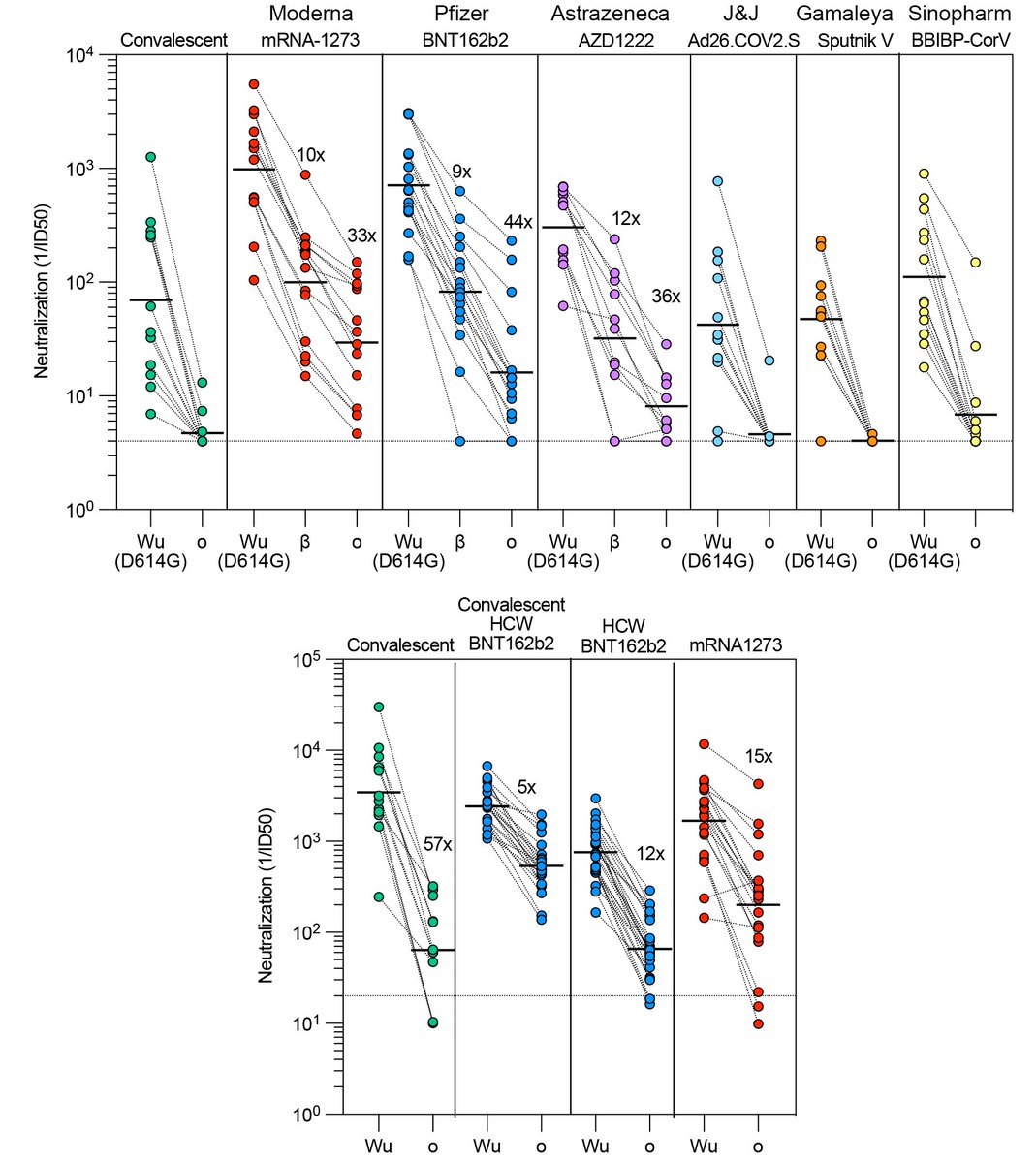
#SARSCoV2 #Omicron immune evasion reduces plasma neutralizing activity up to 57x (convalescent) and up to 30-40x for Pfizer, Moderna or Astrazeneca vaccinees.
No neutralizing activity left with J&J (1x), Sputnik and Sinopharm for most subjects analyzed.
5/8
No neutralizing activity left with J&J (1x), Sputnik and Sinopharm for most subjects analyzed.
5/8
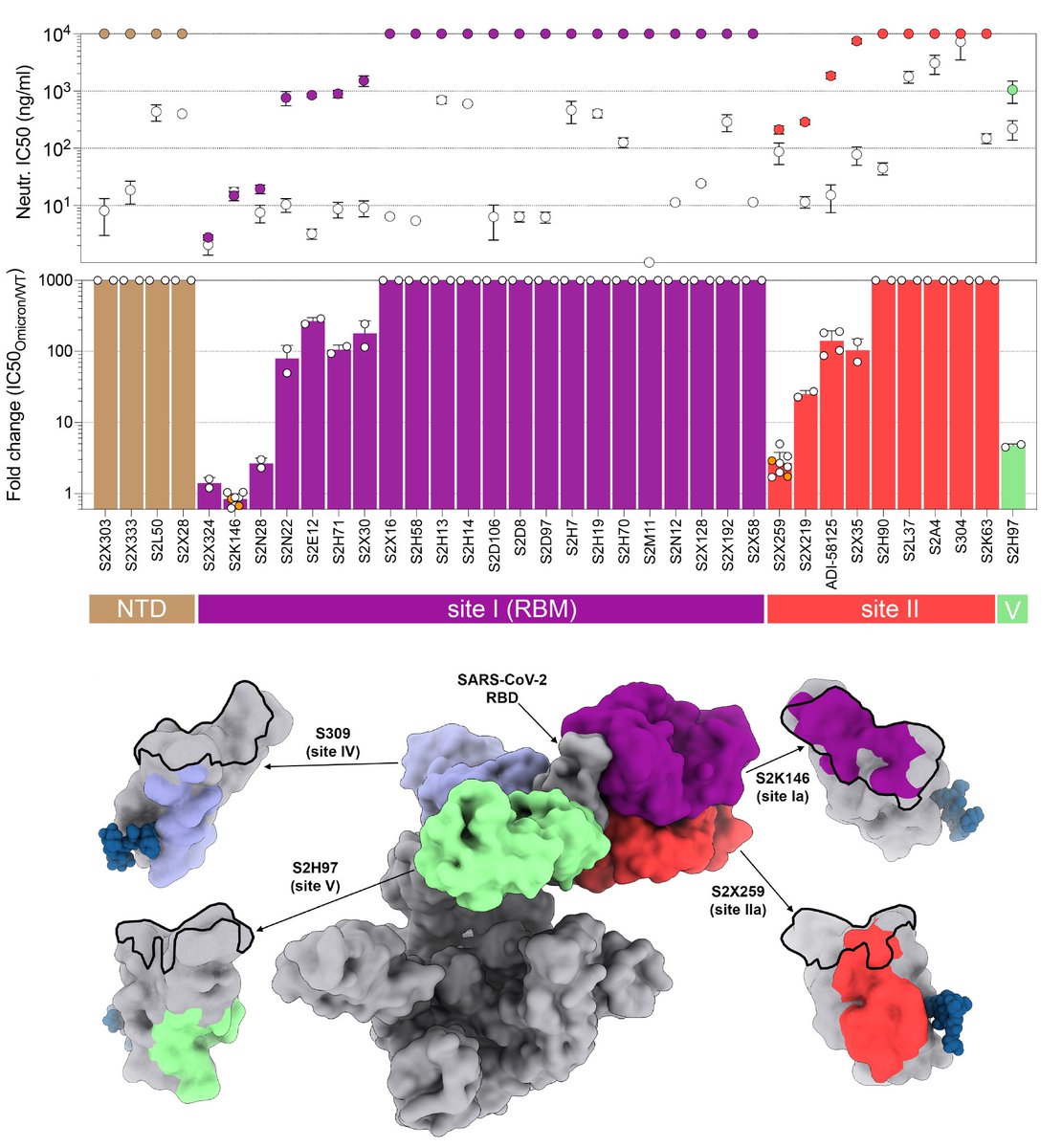
#SARSCoV2 #Omicron mutations knock out 7 out of 8 monoclonal antibodies currently used in the clinic. Only S309, the parent of VIR-7832, retain neutralizing activity, with ~3x reduction relative to Wuhan-Hu-1.
We described S309 early last year.
nature.com/articles/s4158…
6/8
We described S309 early last year.
nature.com/articles/s4158…
6/8
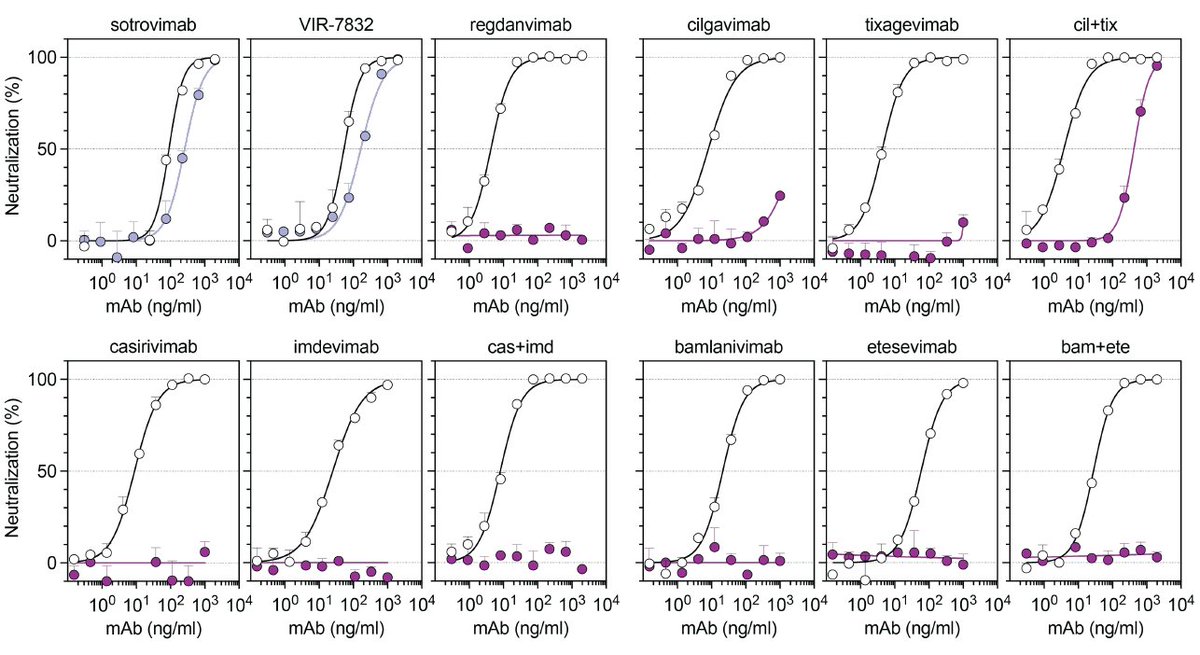
A few other broadly neutralizing sarbecovirus antibodies are inhibiting #SARSCoV2 #Omicron, including S2X259, S2H97 and S2K146. This is good news and shows that targeting conserved sarbecovirus RBD epitopes provides resilience to viral evolution!
7/8
7/8
Thank you so much @SamK_Zepeda Kaiti Sprouse @coronalexington and all our wonderful collaborators including @atelentia @LanzavecchiaB @HelenChuMD @LucaPiccoli9 @SkiClimbSciNick @DrLisaPurcell and many others not on twitter!
8/8
8/8
• • •
Missing some Tweet in this thread? You can try to
force a refresh