From systems to structure — using genetic data to model protein structures go.nature.com/3ncLCWC #Review by @HannesBraberg, @ig_ech, @rmkaake, @salilab_ucsf & @KroganLab
@UCSF @GladstoneInst @QBI_UCSF
@UCSF @GladstoneInst @QBI_UCSF
Recent technologies use large-scale genetic and evolutionary datasets to model the structures of proteins and their complexes 

Coevolution-based methods model protein structures by identifying pairs of amino acid residues that are likely to be close in space because they evolve together

Genetic interaction-based methods use point mutations to identify pairs of residues that share a common function and are likely to be close in space
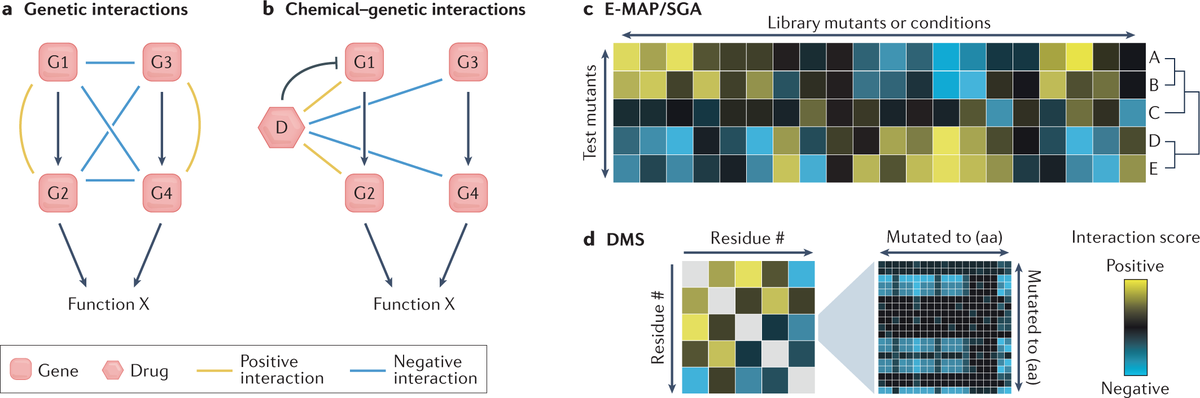
Deep learning is playing an increasingly important role in protein structure modeling and helps extract the most informative content from evolutionary protein sequence data and experimental structures
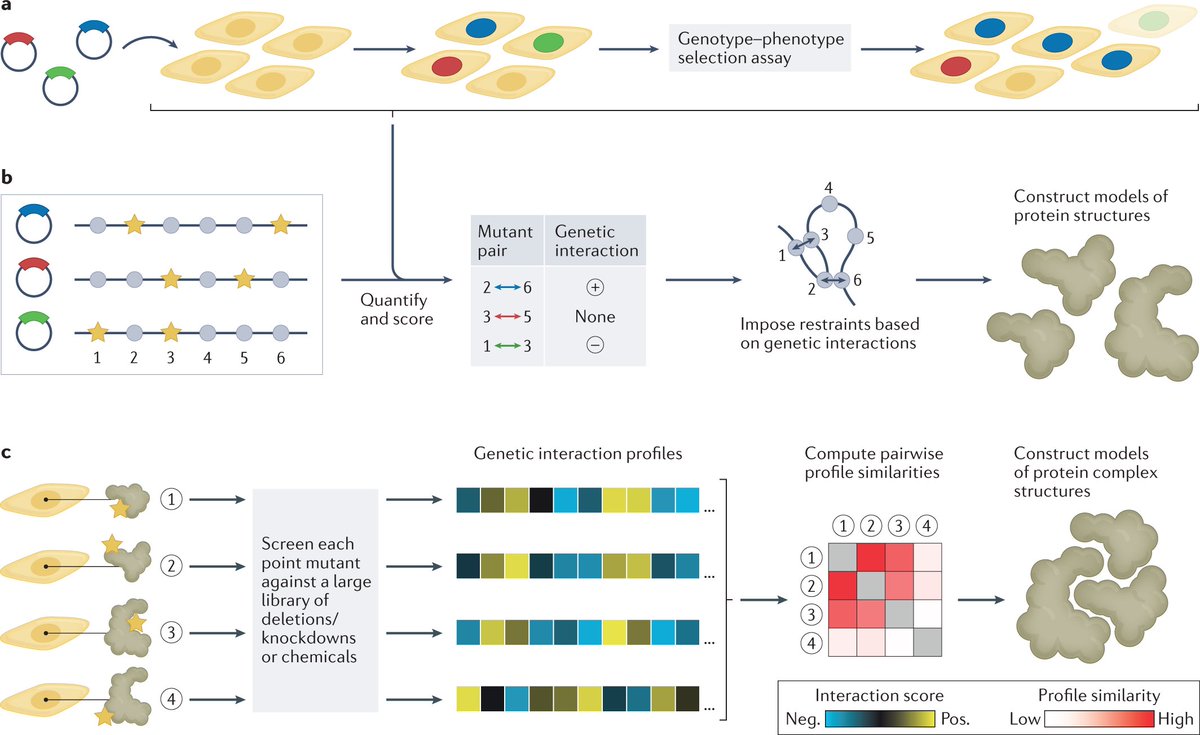
In contrast to traditional structural biology approaches, the discussed technologies rely on data derived from biological function, rather than from physical properties
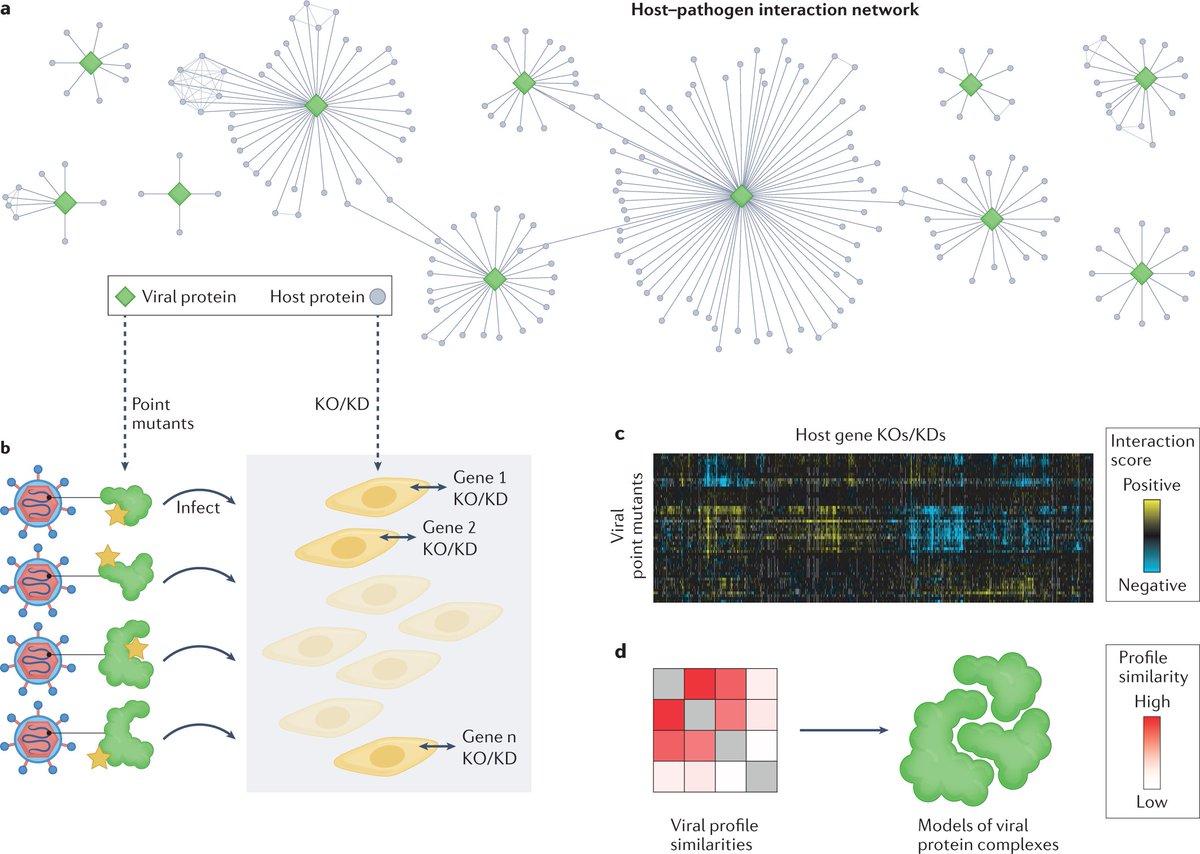
These datasets are primed to complement traditional structural biology methods to provide a more accurate and complete description of the structures of proteins in vivo
• • •
Missing some Tweet in this thread? You can try to
force a refresh