SARS2 and HIV-1 redux
In Feb of 2020, a preprint showing that the SARS2 spike protein has 4 regions of homology with HIV-1 (bit.ly/3rmBtYD) was forced to retract.
So where are these regions in the 3D protein structure?
😲All 4 regions are missing!
Surgically ...
In Feb of 2020, a preprint showing that the SARS2 spike protein has 4 regions of homology with HIV-1 (bit.ly/3rmBtYD) was forced to retract.
So where are these regions in the 3D protein structure?
😲All 4 regions are missing!
Surgically ...
Missing residues usually indicate flexible loops.
So where are these loops? What are they doing?
The first 3 are from the HIV-1 gp120 gene which allows it to bind to host cells containing the CD4 receptor: mostly immune cells like T-cells and macrophages.
O no, what if ...
So where are these loops? What are they doing?
The first 3 are from the HIV-1 gp120 gene which allows it to bind to host cells containing the CD4 receptor: mostly immune cells like T-cells and macrophages.
O no, what if ...
Someone alerted Fauci to the same problem:
bit.ly/3qtU06e
What if the spike protein can interact with the CD4 receptor? Bad news.
Here are the regions in the structure. Spike forms a homo-trimer. The regions (red, yellow, and blue) come together at a peripheral point.
bit.ly/3qtU06e
What if the spike protein can interact with the CD4 receptor? Bad news.
Here are the regions in the structure. Spike forms a homo-trimer. The regions (red, yellow, and blue) come together at a peripheral point.

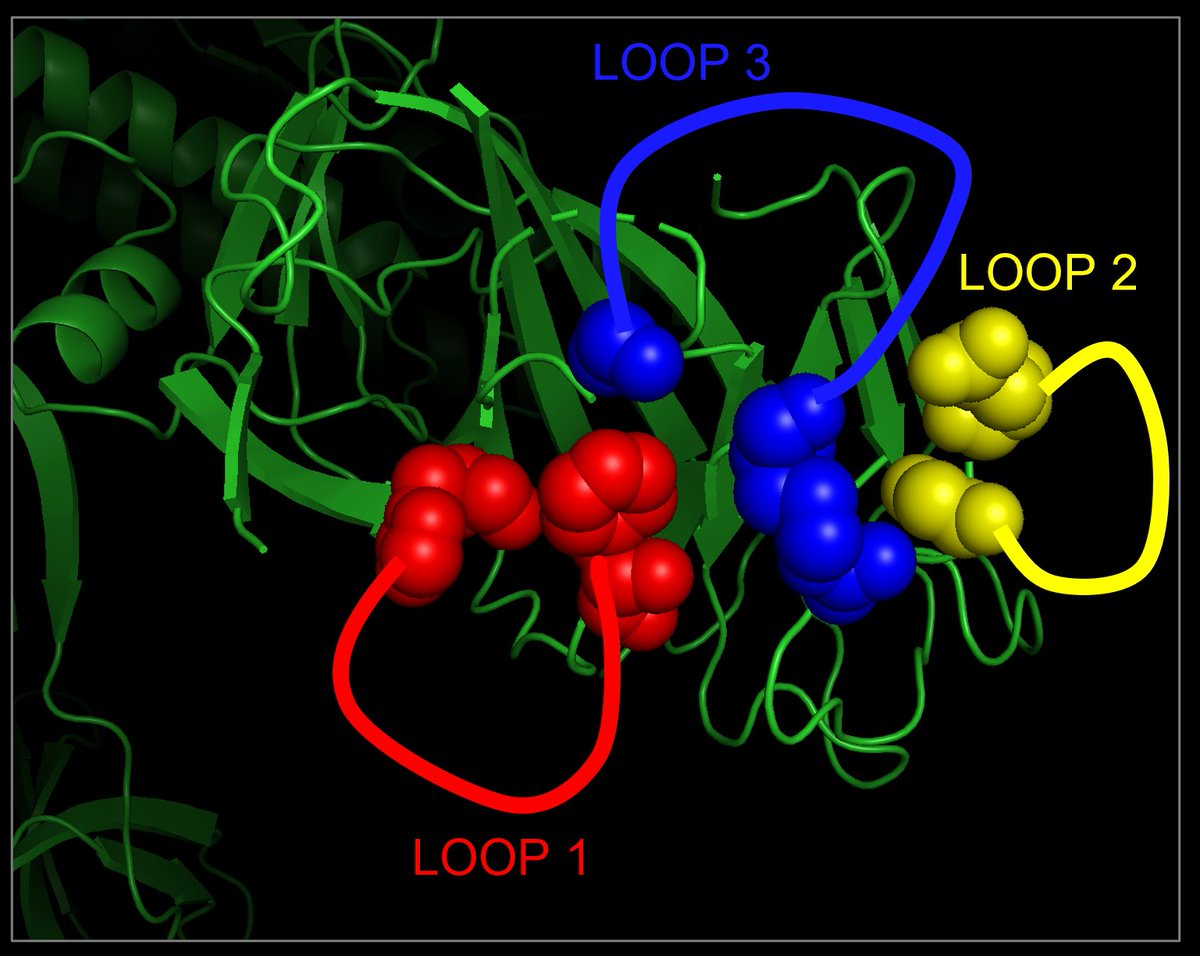
Below is a zoomed-in version for subunit A, with the 3 missing loops drawn in by hand. Note how close their connectors (shown as solid spheres) are in 3D.
So 3 regions with homology to the CD4 binding domain of HIV-1:
- are missing in the structure
- come together in 3D
So 3 regions with homology to the CD4 binding domain of HIV-1:
- are missing in the structure
- come together in 3D
Should we conclude that the SARS2 spike protein can interact with CD4?
Of course not
If the powers-that-be hadn't forced the retraction of the preprint, we would have had an answer by now
Interestingly, #Fauci knew about this in Feb 2020:
bit.ly/3qtU06e
Who alerted?
Of course not
If the powers-that-be hadn't forced the retraction of the preprint, we would have had an answer by now
Interestingly, #Fauci knew about this in Feb 2020:
bit.ly/3qtU06e
Who alerted?
There is already evidence that SARS2 can affect CD4+ T helper cells, lymphocytes that coordinate the adaptive immune response.
Thank you @Rossana38510044 !
bit.ly/3FEuJe0
Thank you @Rossana38510044 !
bit.ly/3FEuJe0
• • •
Missing some Tweet in this thread? You can try to
force a refresh