
1/ Knowing the frequency for alleles of genomic variants in populations around the world helps us understand phenotypes and disease 🌎🌍🌏
We’re here to take you through the data in @ensembl step-by-step. A thread…🧵
#genomics #bioinformatics #tweetorial #Ensembltraining🧬
We’re here to take you through the data in @ensembl step-by-step. A thread…🧵
#genomics #bioinformatics #tweetorial #Ensembltraining🧬

2/ The way you approach this problem will depend on if you are starting with a #gene of interest or if you already have the ID (e.g rs699) of a variant for which you want to find the observed allele frequencies.
3/ If you are starting with a gene, search for the gene name or ID from the #Ensembl homepage and navigate to the Gene tab.
4/ Now, click on ‘Variant table’ in the menu on the left hand side of the page to view all short sequence variants found within the gene.
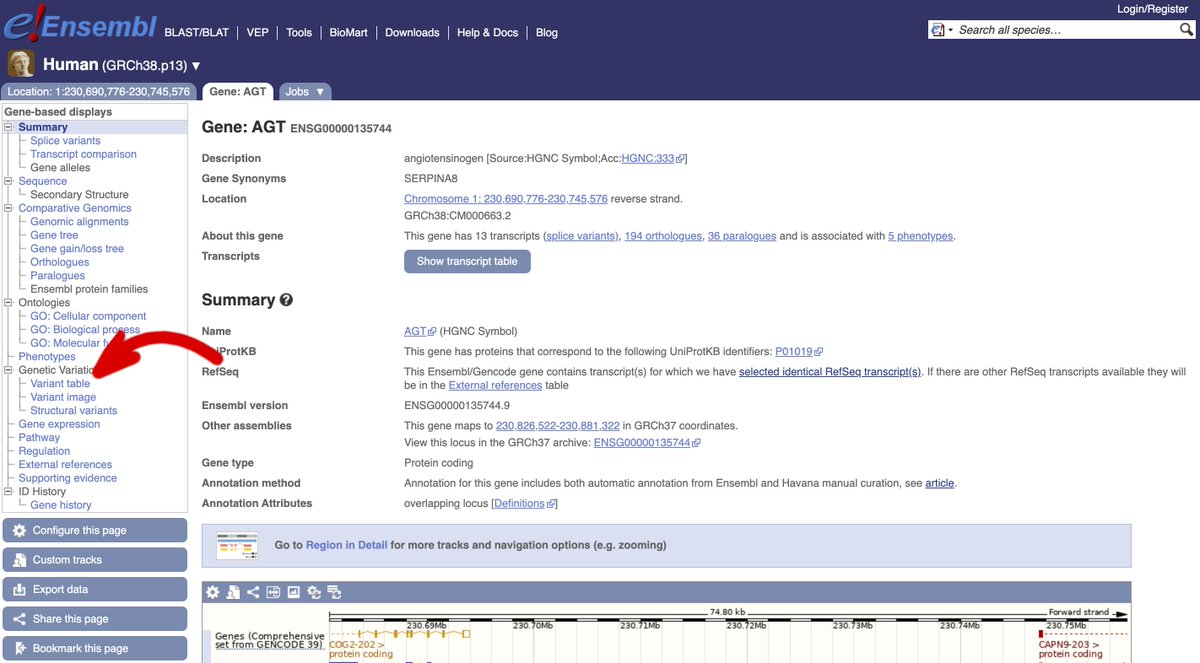
5/ Among lots of other #data, there is a column called ‘Global MAF’. This column will report the Minor Allele Frequency (MAF) observed for this variant in the @1000genomes project, if available.

6/ You can filter the table based on the MAF to show variants with commonly or rarely observed minor alleles. Just click on the blue ‘Global MAF’ button in the Filter section above the table and drag the slider to choose the MAF range to want to see.
7/ ⚠️ Warning ⚠️ This will also remove all variants with no MAF reported from the table.
8/ Now, click on the ID of the variant within the table to navigate to the Variant tab to find out more. Alternatively, if you already know the ID of your variant of interest (e.g rs699), you can search for the variant ID from the homepage.

9/ Now that we’re in the variant tab, click on ‘Population genetics’ to navigate to a page showing observed allele frequencies for different populations across a number of different projects. The data is presented as pie charts and in tabular form as well.
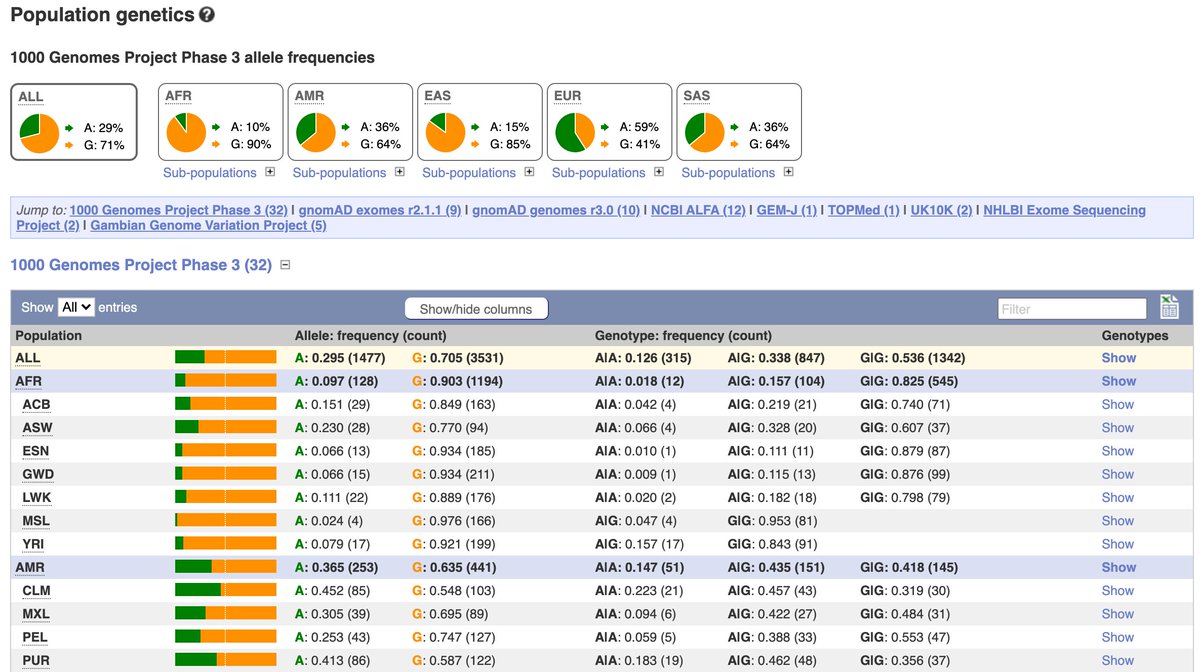
10/ For human 🧑🤝🧑, Ensembl presents allele frequencies from the @1000genomes, @gnomad_project, @NCBI ALFA project, GEM-J project, GGVP, @nih_nhlbi Exome sequencing project, TOPMed and UK10K projects:
ensembl.org/info/genome/va…
ensembl.org/info/genome/va…
11/ Scroll down to see all of the data available for your variant!
12/ Click on ‘Sub-populations’ to see observed allele frequencies in the individual populations that make up the continental populations.

13/ The table shows allele frequencies for each individual population on the left (remember each person sequenced in the projects contributes 2 allele counts as humans are diploid!) and genotype frequencies on the right (homo- or heterozygous)

14/ Click on ‘Show’ in the Genotypes column in the table to jump to a table showing you the observed genotype for each individual. You can use the sample ID to order cell lines derived from each individual in the @1000genomes project from the @Coriell_Science cell repository
15/ There is also allele frequency data for other vertebrates as well as non-vertebrates in @ensemblgenomes:
ensembl.org/info/genome/va…
🐭🐄🐶🐟🌾🦠🦟
ensembl.org/info/genome/va…
🐭🐄🐶🐟🌾🦠🦟

• • •
Missing some Tweet in this thread? You can try to
force a refresh
















