The following paper is one of the most interesting and thoughtful I've read in quite some time.
The authors offer a new framework for understanding if genetic mutations are harmless (benign) or dangerous (pathogenic).
Spoiler: AlphaFold 2 is involved.
nature.com/articles/s4146…
The authors offer a new framework for understanding if genetic mutations are harmless (benign) or dangerous (pathogenic).
Spoiler: AlphaFold 2 is involved.
nature.com/articles/s4146…
First, I'd like to cite the authors; .@capra_lab, .@rodendm, and .@computbiolgeek. I'm sure they'll correct me if I butcher anything.
I originally stumbled on the paper after reading a thread about it by .@RyanDhindsa, which I've linked below:
I originally stumbled on the paper after reading a thread about it by .@RyanDhindsa, which I've linked below:
https://twitter.com/RyanDhindsa/status/1536392983917891585
Some Background:
The basis of medical genetics is understanding how #DNA mutations (variants) give rise to disease.
Recall that inherited DNA sequence variants can sometimes alter proteins by changing the identity of an amino acid.
This is called a 'missense mutation'.
The basis of medical genetics is understanding how #DNA mutations (variants) give rise to disease.
Recall that inherited DNA sequence variants can sometimes alter proteins by changing the identity of an amino acid.
This is called a 'missense mutation'.
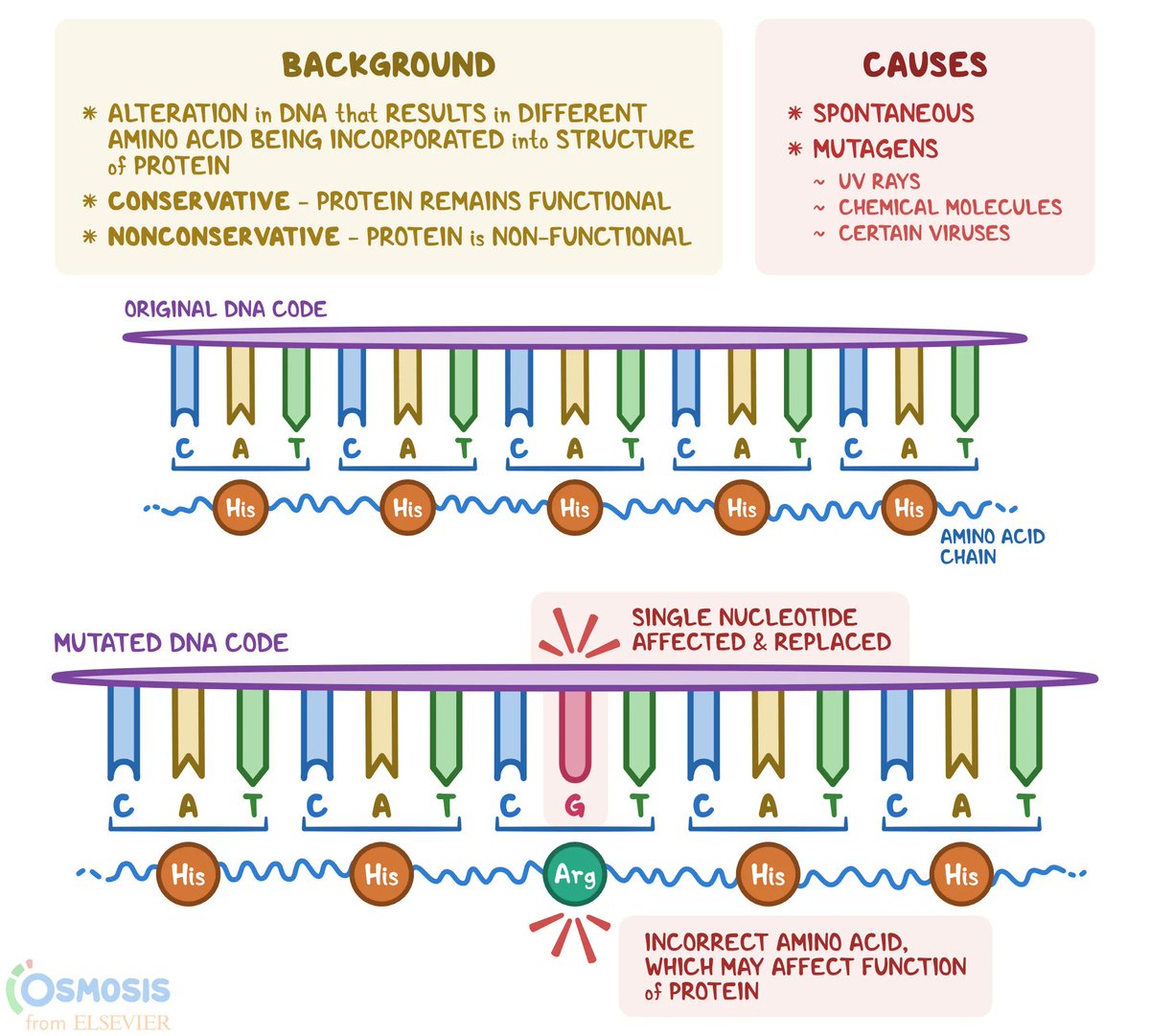
In the above example, the normal (wild-type) DNA sequence reads ...CAT..., which codes for the amino acid histidine. The mutated version reads ...CGT..., which replaces histidine with a different amino acid called arginine.
Recall that proteins = amino acids strung together.
Recall that proteins = amino acids strung together.
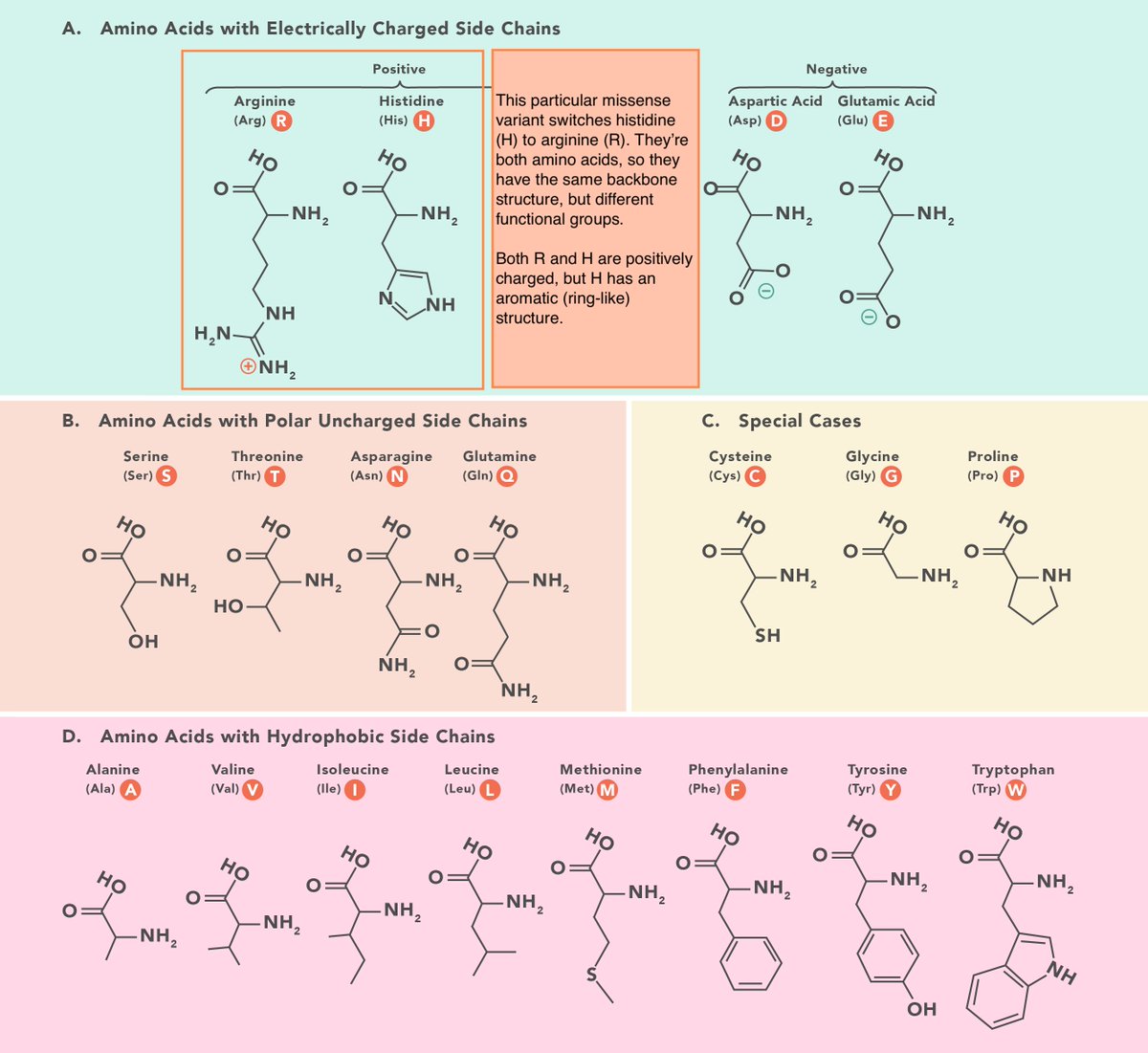
Extrapolating from the example, the million-dollar question is whether a missense variant (and the resulting protein variant) is benign or pathogenic.
In today's age, most single-nucleotide variants (SNVs) we find in patients are variants of uncertain significance (VUSs).
In today's age, most single-nucleotide variants (SNVs) we find in patients are variants of uncertain significance (VUSs).
This means the variant is a bit like Schrödinger's Cat. Until we have enough information to place it in the benign/likely-benign (B/LB) or pathogenic/likely pathogenic (P/LP) bins, it could be either.
Fortunately, most VUSs are downgraded to B/LB.
ncbi.nlm.nih.gov/pmc/articles/P…
Fortunately, most VUSs are downgraded to B/LB.
ncbi.nlm.nih.gov/pmc/articles/P…
To label a variant along the pathogenicity spectrum, large clinical labs use an ensemble of methods ranging from a simple literature review (who's seen this before) to physically recreating the mutation in a lab experiment to see what happens.
gimjournal.org/article/S1098-…
gimjournal.org/article/S1098-…
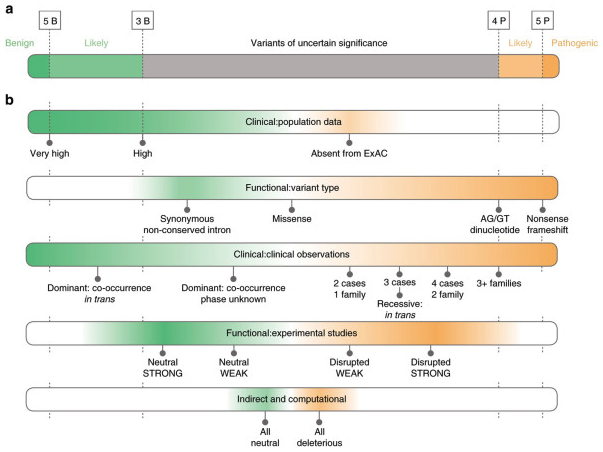
Once they confidently interpret a variant as benign or pathogenic, labs often submit this 'truth label' to open-source databases (e.g. ClinVar), which other labs can reference if they happen to find the same mutation in one of their patients.
https://twitter.com/sbarnettARK/status/1528812202865709064
Still, think about how large sequence space is. Every one of us has ~6 billion base pairs of DNA in our cells. The exact letter change, the location of the letter change, and the superposition of many letter changes in concert can create enormous complexity.
SNVs are also only one type of variant - there are insertions/deletions (indels), large structural variations, and many other types that are beyond the scope of this thread.
Sufficed to say - we need better tools to find if SNVs are dangerous, even if not in a database.
Sufficed to say - we need better tools to find if SNVs are dangerous, even if not in a database.
Before describing their new method (COSMIS), the authors walk through several contemporary approaches to interpret SNVs.
1. Interspecies population frequency = Is the sequence conserved in related species across long timespans? If so, it's important that it stay unmutated.
1. Interspecies population frequency = Is the sequence conserved in related species across long timespans? If so, it's important that it stay unmutated.

2. Intraspecies population frequency = In humans, is the variant extremely rare? If so, it's probably dangerous to carry because evolution selected against it. This reminds me a bit of the 'survivorship bias' plane.
deanyeong.com/article/surviv…
deanyeong.com/article/surviv…
Still, population-based measures look at correlation only. They don't tell you anything about HOW a protein is altered by an upstream DNA sequence change. Nor do they give insight into how a protein variant then gives rise to a disease state.
3. Some newer, functionally-informed metrics overcome the limitations of statistical approaches but may be low resolution. That is, they can say if a gene or region of a gene is essential, but don't go down to the amino-acid level.

Many of these tools consider the 1D sequences of genes or proteins but don't necessarily consider that while these molecules can be represented as a string of characters - they are, in fact, dynamic 3D entities that interact with their environments.
As shown below, the authors' framework (COSMIS) analyzes missense-mutated proteins in 3D. Here, they define a 'contact set' as amino acids that are physically close in 3D space.
Notice that j2 and j5 are close in 1D (sequence) space, but don't interact in 3D (real) space.
Notice that j2 and j5 are close in 1D (sequence) space, but don't interact in 3D (real) space.

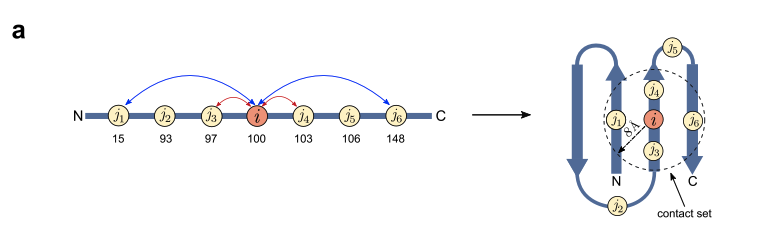
Why does that matter?
Remember earlier we talked about a missense variant switching two (+) charged amino acids (H>R)?
Consider what would happen if the switch was to a (-) charged acid. The opposite charges may attract, perhaps misfolding it in a destructive way.
Remember earlier we talked about a missense variant switching two (+) charged amino acids (H>R)?
Consider what would happen if the switch was to a (-) charged acid. The opposite charges may attract, perhaps misfolding it in a destructive way.
Conceptually, the authors took millions of known SNVs, mapped those variants to (1D) protein sequences, and then overlaid those on 3D protein structures.
Some structures were experimentally determined, but many were simulated using .@DeepMind's AlphaFold 2 (AF2).
Some structures were experimentally determined, but many were simulated using .@DeepMind's AlphaFold 2 (AF2).
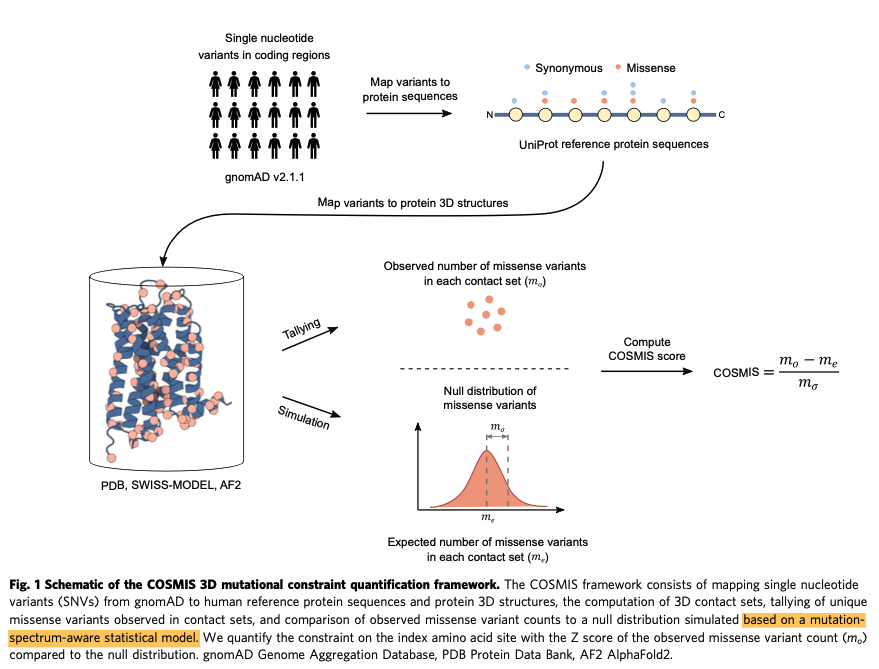
By subtracting the # of observed missense variants from the expected # (and dividing by the standard deviation of the expected distribution), the authors created the COSMIS score.
The lower it is, the more likely a missense variant is pathogenic (dangerous).
The lower it is, the more likely a missense variant is pathogenic (dangerous).
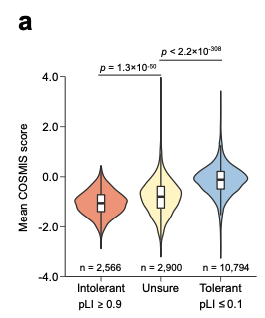
Not only does COSMIS outperform other 1D and 3D-aware variant interpretation metrics, but it carries complementary information to phylogenetic (population) models, as shown in (a) and (c) below, respectively.
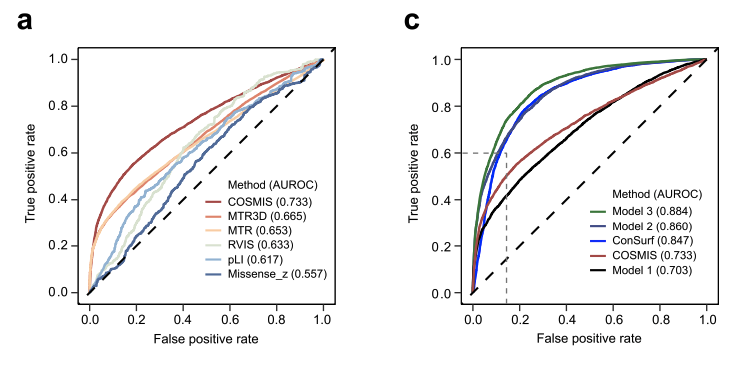
Altogether, this work shows how thinking in 3D helps scientists and clinicians interpret the functional consequences of DNA sequence changes.
Yes, DNA, RNA, and proteins are information, but they're also fragile, physical things.
Yes, DNA, RNA, and proteins are information, but they're also fragile, physical things.
Each of the 20 amino acids is unique in size, shape, charge, and other qualities. By considering how they interact in 3D space, COSMIS better predicts how the overall function of a protein changes.
While not a panacea, it's a great step towards linking sequence and effect.
While not a panacea, it's a great step towards linking sequence and effect.
Unroll @threadreaderapp
• • •
Missing some Tweet in this thread? You can try to
force a refresh