
1/ My favourite gene has 15 transcripts, which one should I use for further analysis? To report the position of variants? To design primers for? 🤯
This #tweetorial will show you how to filter and prioritise transcripts… 🧵
#genomics #bioinformatics #Ensembltraining 🧬
This #tweetorial will show you how to filter and prioritise transcripts… 🧵
#genomics #bioinformatics #Ensembltraining 🧬

2/ To start, look for the #canonical tag in the flags column of the transcript table. The canonical transcript is based on conservation, expression, concordance with @appris_cnio and @uniprot, length, clinically important variants and completeness.

3/ Many Ensembl #canonical transcripts will also be the #MANESelect, which is our collaboration with @NCBI. These transcripts match perfectly with RefSeq transcripts, so are the best to report variant location.

4/ Some genes have mutually exclusive exons which are clinically important. These genes may also have a #MANEPlusClinical, a second transcript with the other exon, and you should report variant positions in both.
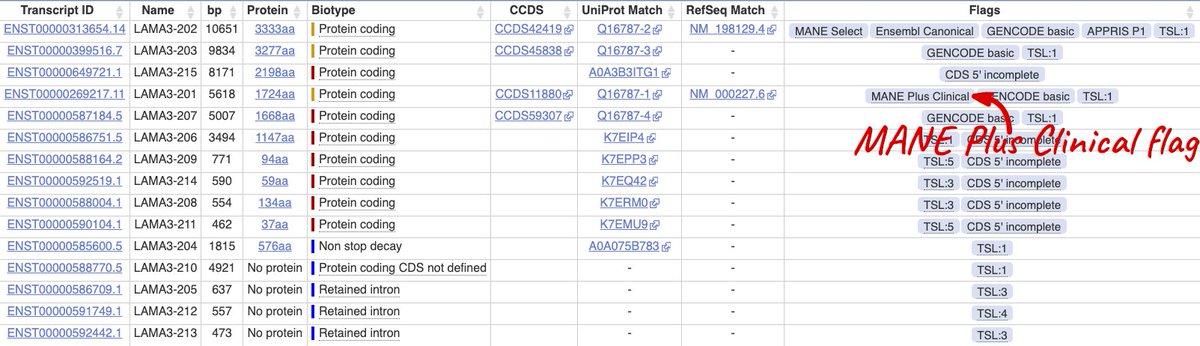

5/ ⚠️WARNING ⚠️ The MANE project has only focussed on human protein coding genes on the human GRCh38 assembly, so MANE Select or MANE Plus Clinical transcripts are only annotated for this assembly.
6/ ⚠️WARNING ⚠️: Don’t use the transcript numbers (eg LAMA3-201) or transcripts IDs (eg ENST00000269217) as rankings or a guide for choosing. Both are completely arbitrary and only the ENSTs are stable between releases.

7/ Whatever you use to report, note down the transcript stable ID with the version number, eg ENST00000313654.14. Transcripts change and can differ between databases. Only way to be 100% clear about which sequence is with a versioned stable identifier.
• • •
Missing some Tweet in this thread? You can try to
force a refresh
















