Multi-Omic inTegrative Analysis (MOTA): an application of differential network analysis to #multiomics.
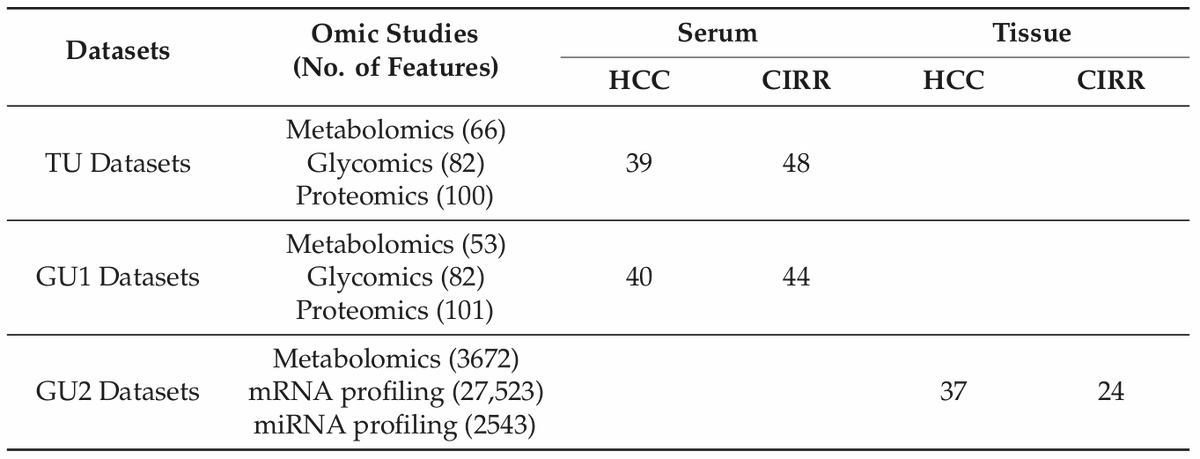
✓3 non-TCGA datasets (HCC vs CIRR)
✓explanations for parameters & rgCCA choice
✓good at known cancer drivers recovery
✓consistent across cohorts
#SundayMultiOmics 1/n
✓3 non-TCGA datasets (HCC vs CIRR)
✓explanations for parameters & rgCCA choice
✓good at known cancer drivers recovery
✓consistent across cohorts
#SundayMultiOmics 1/n
[[ Detailed description & comments follow ]]
Link: doi.org/10.3390/metabo…
Figures © by authors, reused under CC-BY 4.0
creativecommons.org/licenses/by/4.…
Developed @LombardiCancer @gumedcenter (sorry I could not to find authors on Twitter)
Link: doi.org/10.3390/metabo…
Figures © by authors, reused under CC-BY 4.0
creativecommons.org/licenses/by/4.…
Developed @LombardiCancer @gumedcenter (sorry I could not to find authors on Twitter)
["Layman" introduction]: many studies focus on the differences in the abundance of specific biomolecules; while very useful to highlight the obvious #biomarkers, it may not explain complex disease mechanisms & can be very sensitive to cohort differences (e.g. in #metabolomics)
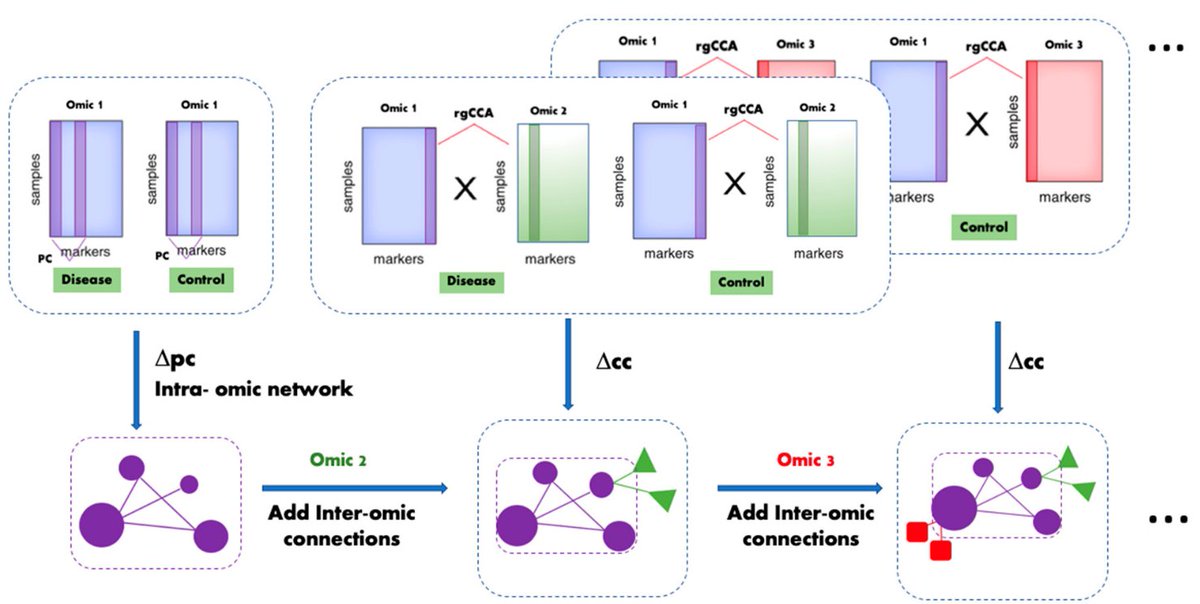
So, what if multiple studies propose very different biomarkers for the same disease? Meta-analysis is not the only way out. Comparing correlation networks between patients and controls can help distinguish the true markers from the noise:
it is one thing to show that a miRNA has a different abundance between patients, it is another to show that it translates to a tangible impact on thousands of genes. This example highlights that we cannot restrict analyses to a single omic: regulation often happens between omics!
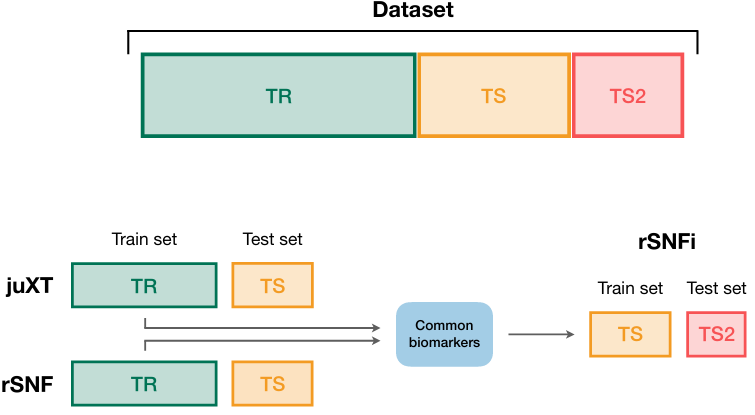
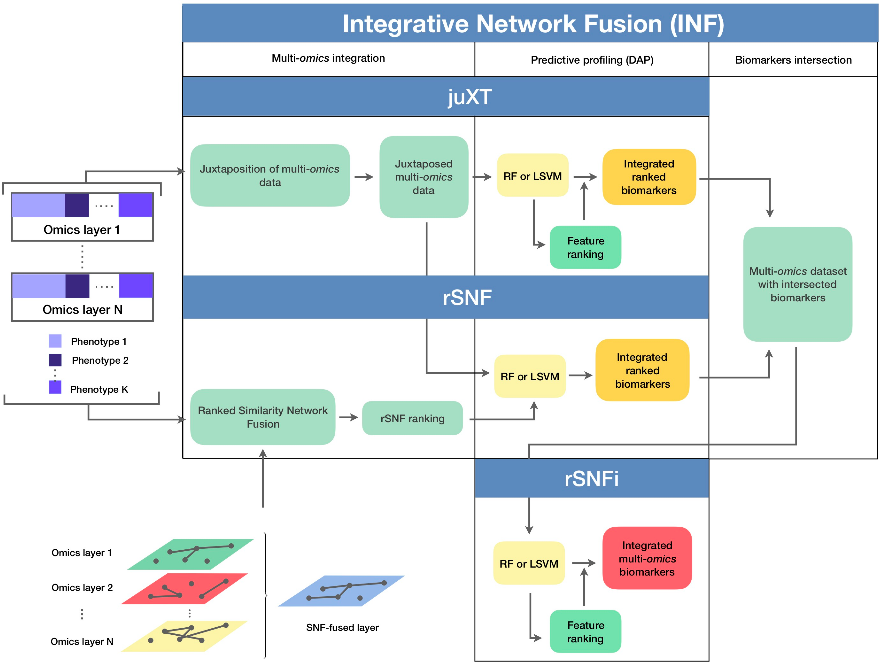
[MOTA] This method performs "asymmetric" omics network integration; it might not be universal, but seems great if you aim to investigate biomarkers for one specific omic (e.g. metabolites) & then build up additional confidence/improve prioritization with use of additional omics.
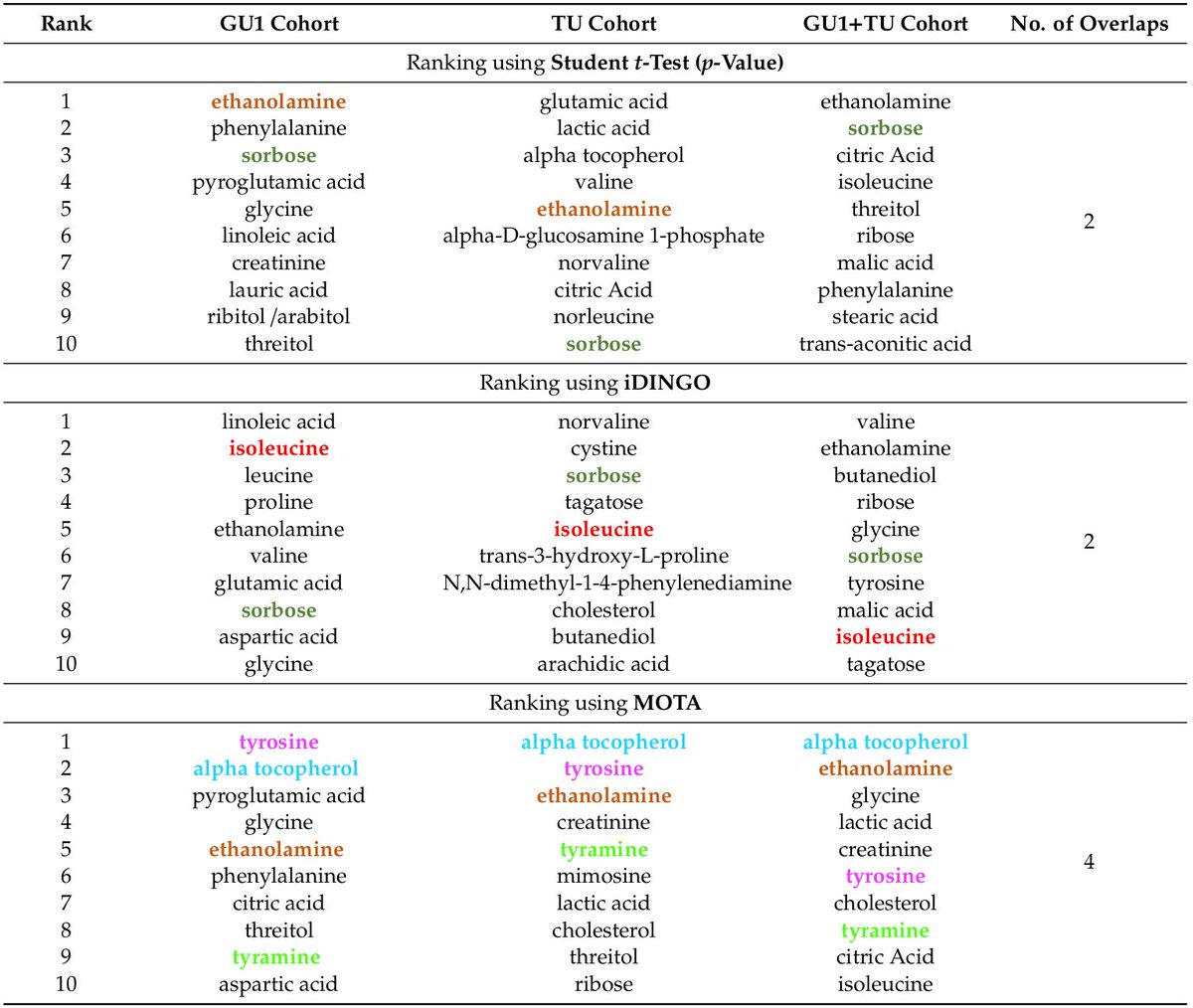
Each node is assigned a p-value using a differential expression method (e.g. t-test, but possibly could be DESeq or limma for #transcriptomic layer). The score of a node is derived from this p-value and p-values of nodes connected to it (aggregated by omic with Stouffer’s method)
I call it "asymmetric" omics analysis, because rather than investigate connections between all omics, one of them (the first one) is "singled out": all "ancillary" omics are added on top of it. Authors propose to investigate all-vs-all scenario in future works.
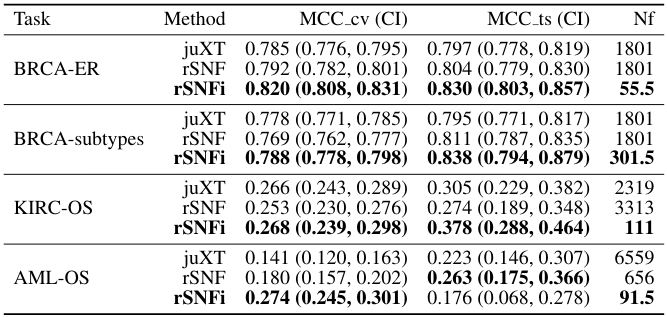
[Results] Authors show that MOTA provides more consistent results across cohorts (than t-test and iDINGO) and recovers more known cancer driver (COSMIC) genes (than #DESeq2 and iDINGO). It's outside of my expertise to comment on the obtained metabolite&gene lists in HCC context.
[Comments] It's great to see a method evaluated on three original datasets (which provide nice support for its utility). It would be even better, if the standard TCGA datasets were included, to enables some sort of between-study comparison (esp. in the absence of the source code)
I am saddened to see such a promising method published without any source code, but I do hope that the authors are working towards releasing it along with the future works (which sound interesting!).
A limitation of many #multiomics papers is a small number of comparisons against existing methods; it was already a problem in of bioinformatics: it's time-consuming and risky (what if your method is worse?), but is sometimes taken to a whole new level in multi-omics; indeed, ...
... dealing with multiple omics is more challenging and lack of standardised APIs (or code source at all) & limited number of datasets does not help. Is this reasonable to propose that we should at least compare to naive methods, such as log. LASSO/PLS-DA on concatenated omics?
Finally, I understand that sharing data from human studies may not be easy, but it would be great if the synthetic datasets (even simple simulations, but ideally reflecting the characteristics of the datasets used in the study, like Simulacrum but for molecular data) were shared.
Bonus: the use of permutation testing may be a strength of the method, but as always comes at a significant computational cost; for the larger dataset (which is still small as for today's standard) it took ~1 hour to complete on a modern Mac (not great, but not very bad either).
@ThreadReaderApp unroll