
A true gem among #multiomics preprints: Integrative Network Fusion by @MarcoChierici, @nicole_bussola, @viperale, et al:
✓ 3 TCGA cancers & simulated data
✓ cross-validation described in detail
✓ flow diagram
✓ source code & data shared
✓ packages w/ version, cited
/n
✓ 3 TCGA cancers & simulated data
✓ cross-validation described in detail
✓ flow diagram
✓ source code & data shared
✓ packages w/ version, cited
/n

- [the method description & comments follows]
- link: biorxiv.org/content/10.110…
- licence the above figures/tables: CC BY-NC-ND 4.0
- an earlier version of INF was previously presented in 2018: doi.org/10.1186/s13062…
- this is the first tweet in #SundayMultiOmics series
- link: biorxiv.org/content/10.110…
- licence the above figures/tables: CC BY-NC-ND 4.0
- an earlier version of INF was previously presented in 2018: doi.org/10.1186/s13062…
- this is the first tweet in #SundayMultiOmics series
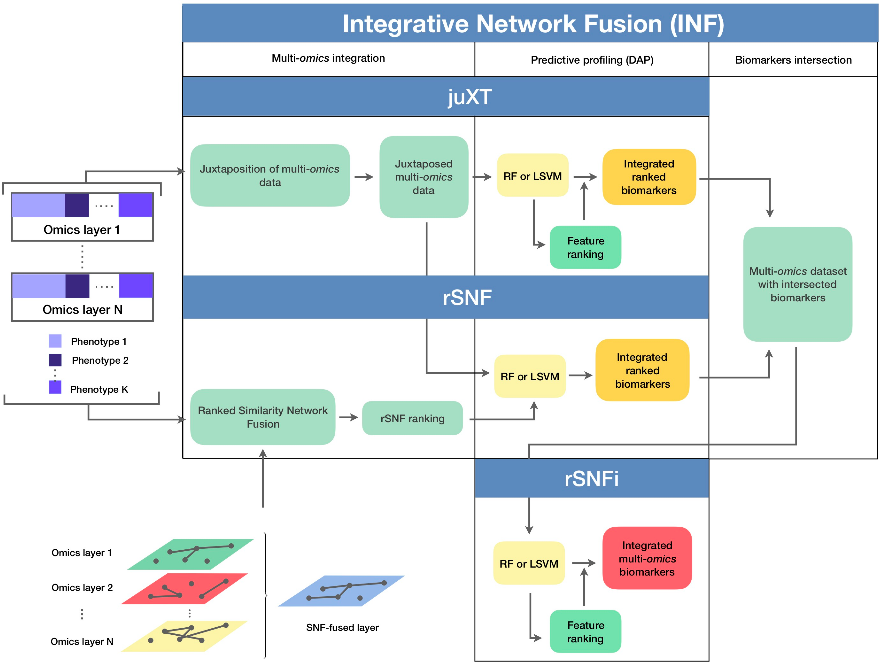
[[Introduction]]: Similarity network fusion (SNF, doi.org/10.1038/nmeth.…) is a popular technique (600+ citations, a lot for multi-omics!) for getting a sort of consensus signal from multiple omics; it requires the same patients (less commonly - observations) in each omic.
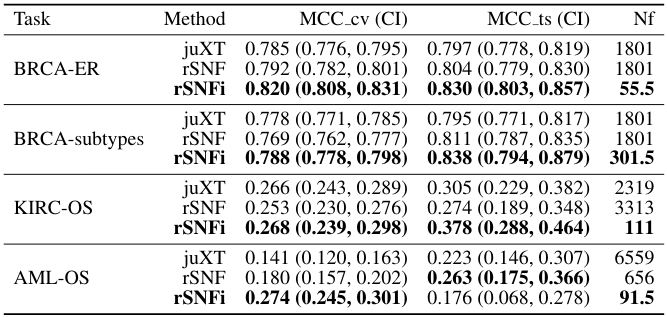
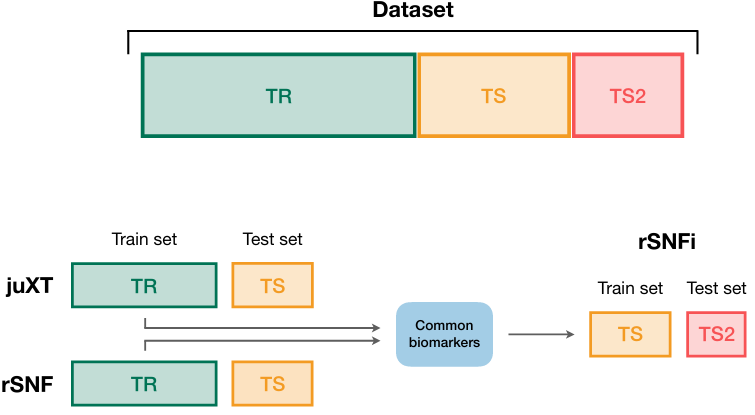
Integrative Network Fusion (INF) uses SNF followed by random forest (RF), or support vector machines (SVM), as well as a more conventional paired omics modelling (also followed by RF/SVM) to extract two sets of candidate features; the overlap is then used in the final model (RF).
The features from the paired omics are extracted with what they call "variable juxtaposition (juXT)"; I am not convinced by this term, but it seems to overlap with early integration approaches (with the simplest form being omics matrices concatenation).
[[Outcomes in prediction]]
- breast invasive carcinoma: ER status, subtypes
- acute myeloid leukemia: overall survival
- renal clear cell carcinoma: overall survival
Comment: BRCA subtypes used (PAM50) are defined by transcriptomic signatures; as such the multi-omic methods...
- breast invasive carcinoma: ER status, subtypes
- acute myeloid leukemia: overall survival
- renal clear cell carcinoma: overall survival
Comment: BRCA subtypes used (PAM50) are defined by transcriptomic signatures; as such the multi-omic methods...
...(other than using RNA + protein) can be disadvantaged. There is some literature suggesting that we should look at these subtypes as "molecular events" (patient can have more than one); some even question their utility as the gold standard. Authors may want to discuss this :)
[[Omics]]
- breast invasive carcinoma: #transcriptomics (mRNA), #proteomics (I guess RPPA), genomic data (copy number variation, #CNV)
- AML & KIRC: transcriptomics: mRNA & miRNA + #methylation
Note: an explanation of why those particular cancers/omics were chosen would be nice!
- breast invasive carcinoma: #transcriptomics (mRNA), #proteomics (I guess RPPA), genomic data (copy number variation, #CNV)
- AML & KIRC: transcriptomics: mRNA & miRNA + #methylation
Note: an explanation of why those particular cancers/omics were chosen would be nice!
[[Cross-validation]]: 10 times repeated 5-fold CV is used; the 10 repeats give us confidence that it was not just one "lucky" result. However, I would love to see 10-fold and 3-fold CV as well - this would tell us how this method behaves when given more (or less) training data...
which is important as it will both inform how much patients we need to get a benefit from measuring multiple omics and whether this method is fit for smaller-scale experiments.
It seems that the accelerated version intentionally "leaks" data in the CV step...
It seems that the accelerated version intentionally "leaks" data in the CV step...
...as the SNF is performed only once, on the full train set (rather than in CV), but this should not affect the overall validity as a test set was set aside beforehand.
[[Evaluation]]: Authors use Matthews Correlation Coefficient which is said to have an advantage over F1 score, as it takes the class imbalance into account. As with F1, we would ideally want to see the sensitivity/specificity (or similar) breakdown to get a full picture.
[[Data]]: in a perfect world, results for all cancers would be included in the supplement (but knowing the TCGA dataset, the three they used might be the only complete enough); the synthetic data can be reproduced, as authors provided the random seed and environment details (✓)
[[Comment on the materials shared]] Authors shared the source code with the full revisions history (git), which is a higher standard compared to just uploading a zip. They also documented the repository and provided instructions allowing for the code reuse...
the code is moderately commented, well-formatted and includes a #jupyter notebook (IMO, would be better to preserve outputs!). Also, it's a good use case for #conda and #snakemake. The input data was shared on figshare (maybe adding the synthetic data would be even better?).
[[SNF comments]] I have previously found that spectral clustering may not be optimal, but I understand authors choice not to play around with the SNF definition. Similarly, one could question whether NMI is the best metric (I believe it's not).
[[Presentation comments]] It took me a while to understand why authors say "RF or SVM"; I think they simply mean that they go with RF by default but also tried SVM and it had a comparable performance (& provide data in supplement) - a clarification earlier in the text could help!
[[Summary]] This is a great work and I would like to see it published in a journal it deserves. While it certainly could be improved in many ways, I wish all the #MultiOmics papers followed at least the same standard. The future work outlook (+histology&radiology) is fascinating!
For the full list of authors on Twitter see:
/n
/n
[[Bonus: the question of baseline]] Both matrix concatenation (the most naive early integration approach) and SNF can be used to boost signal consistent across omics (which would be chopped of in a thresholding step if we used a late integration approach)...
...however, the matrix concatenation is troubled by collinearity and S/N ratio issues, thus many simple methods (such as lasso logistic regression on concatenated matrices) may struggle. I do not know if the RF/SVM is good enough baseline to give a solid base for comparison, but.
...as many papers in the ML field fail to optimize hyperparameters of the method they claim to be superior to, we need to be vigilant in the multi-omics field to compare to the gold standard, and not to a convenient naive approach; to do so we need more up-to-date benchmarks, ...
...and as authors note it is currently unfeasible to get a fair comparison of different methods, and IMO we need a continuous benchmarking platform (we could draw inspirations from Kaggle) with sustainable funding to understand the landscape of the #multiomics methods.
Also, @mikelove's github.com/mikelove/aweso… got a mention. Is the community shifting towards citing repositories, or is it going to stay a rare sight?










