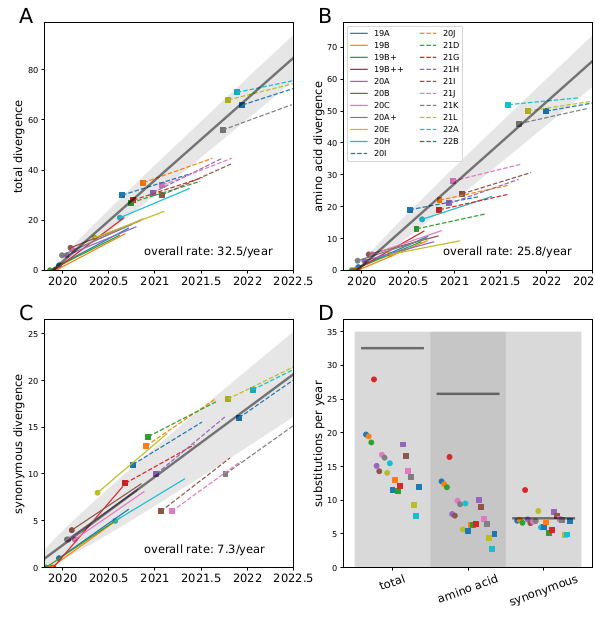
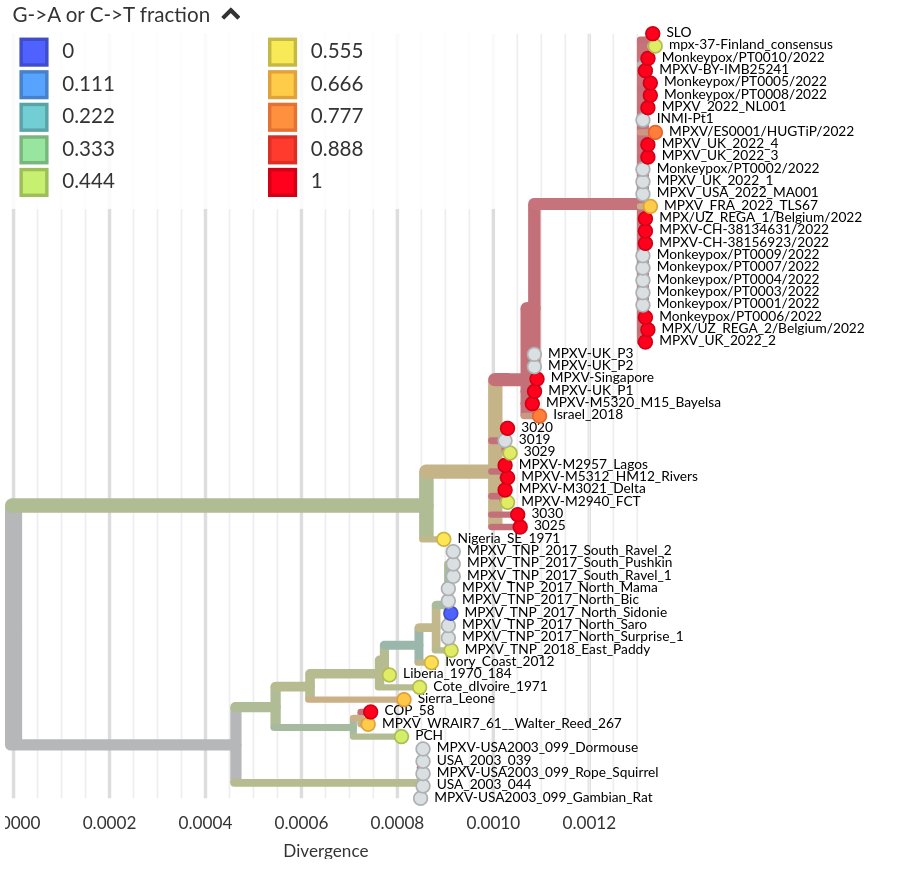
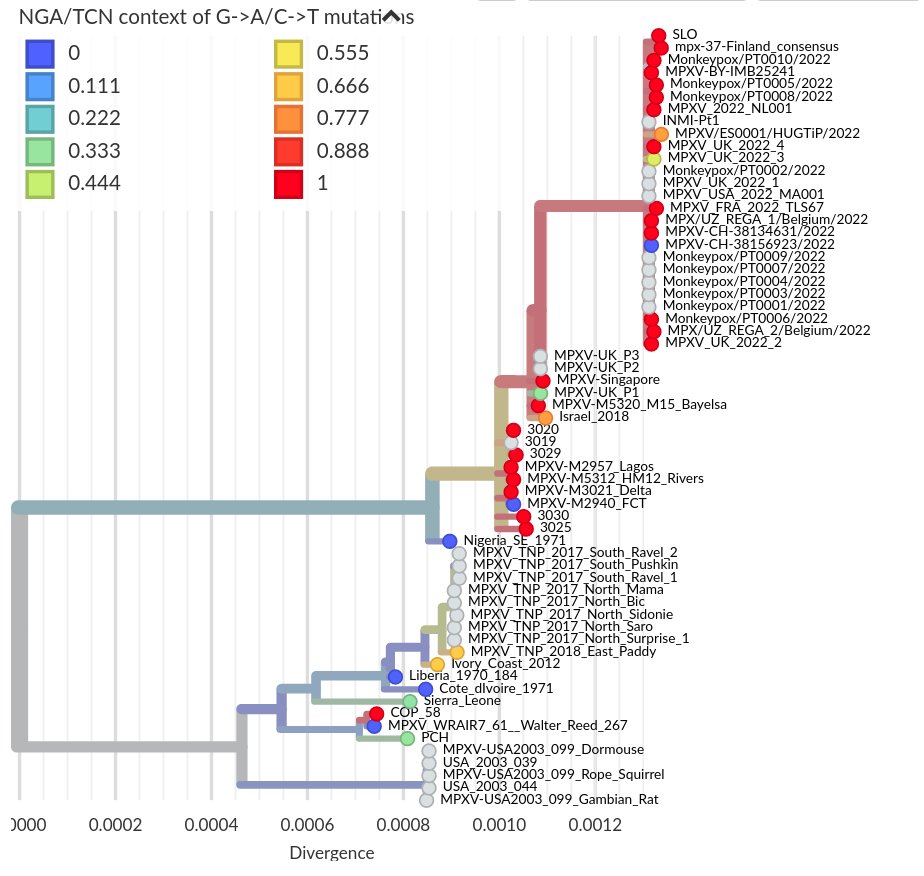
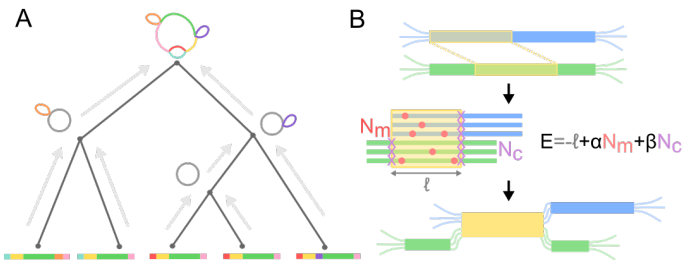
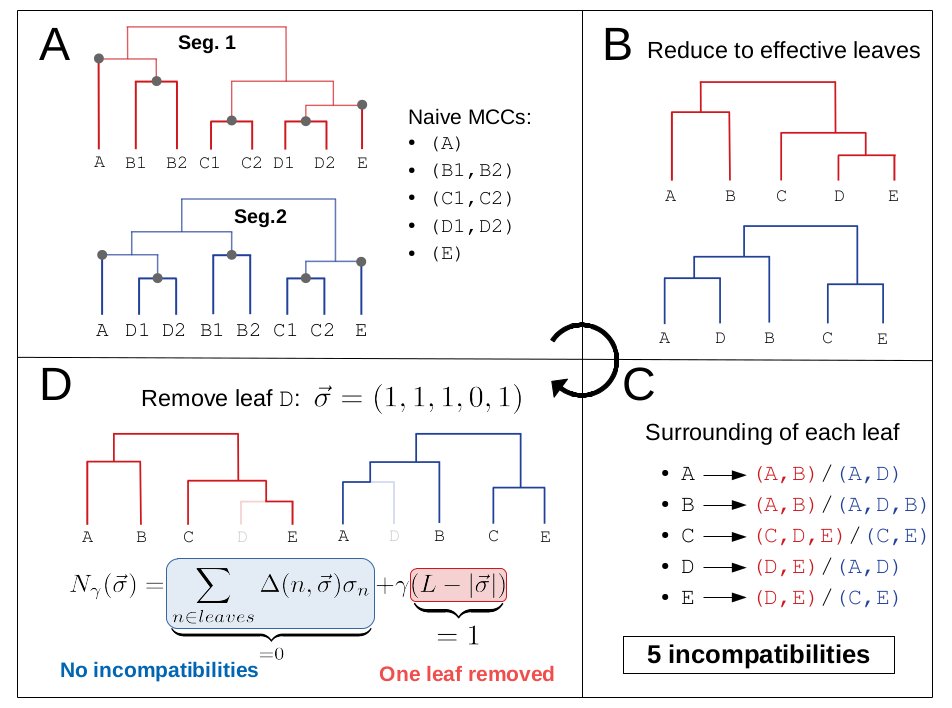
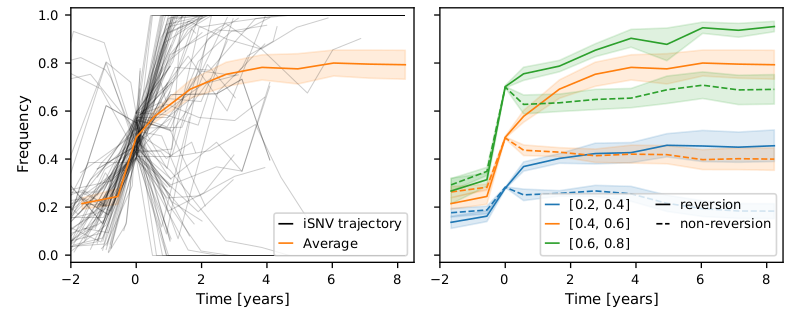
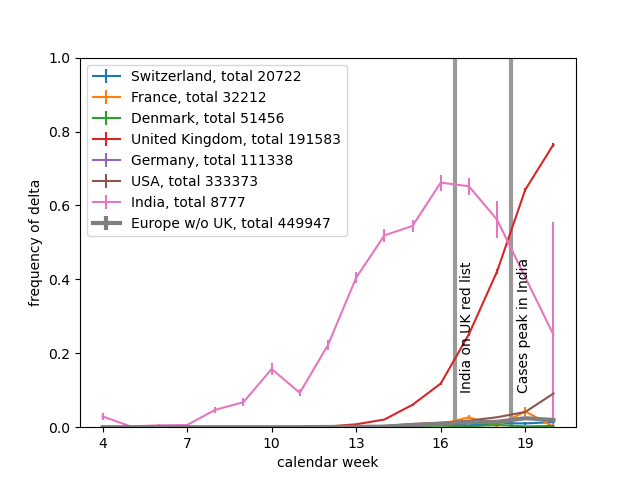
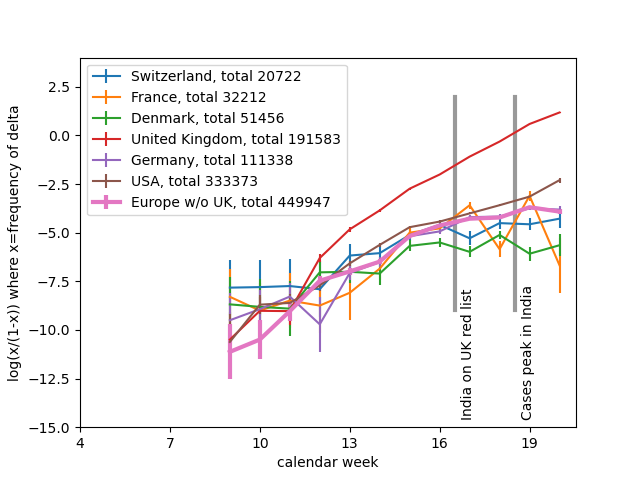
I think the least disruptive way to control #SARSCoV2 is to act early, focus on high transmission settings, masks, and Test-Trace-Isolate-Quarantine to keep Re below 1. But with rising cases, this strategy looks less and less plausible in many parts of Europe.
[1/7]
[1/7]
This could be a prelude to what I fear will be a difficult winter. Controlling #COVID19 will require combinations of tighter measures with difficult trade-offs and serious societal, health, and economic implications.
[2/7]
https://twitter.com/richardneher/status/1293857065425866754
[2/7]
When implementing economic stimuli, we have to keep in mind that the pandemic is not the only crisis we face:
1) The pandemic will be over in 2021 or 2022 (widespread immunity, hopefully via a vaccine).
2) The climate crisis is here to stay.
[3/7]
1) The pandemic will be over in 2021 or 2022 (widespread immunity, hopefully via a vaccine).
2) The climate crisis is here to stay.
https://twitter.com/rahmstorf/status/1220699044181368838
[3/7]
Furthermore:
The old are much more likely to suffer or die from #COVID19, while the young bear the lion share of the cost of restrictions.
Previous and current generations benefited from burning fossil fuels, while the young will suffer most from the climate crisis.
[4/7]
The old are much more likely to suffer or die from #COVID19, while the young bear the lion share of the cost of restrictions.
Previous and current generations benefited from burning fossil fuels, while the young will suffer most from the climate crisis.
[4/7]
Instead of ballooning the stock market and propping up moribund industries, I feel strongly that any economic stimulus needs to be targeted at providing opportunities for future generations and at protecting the climate.
labour.org.nz/release-renewa…
[5/7]
labour.org.nz/release-renewa…
[5/7]
A firm commitment to address the challenges of the future, along with clear communication of #COVID19 risks and transmission patterns, would go a long way to convince society as a whole to control the virus and make a short-term sacrifice worthwhile.
[6/7]
[6/7]
On the other hand, shaming particular groups for spreading the virus is completely unacceptable. Blame and stigmatization have consistently hampered control of many other infectious diseases, #COVID19 is no different.
[7/7]
[7/7]
PS: I am no longer as young as in my profile pic.
• • •
Missing some Tweet in this thread? You can try to
force a refresh