
SARS-CoV-2 surveillance by @my_helix update
- New dashboard: you can check all lineages now
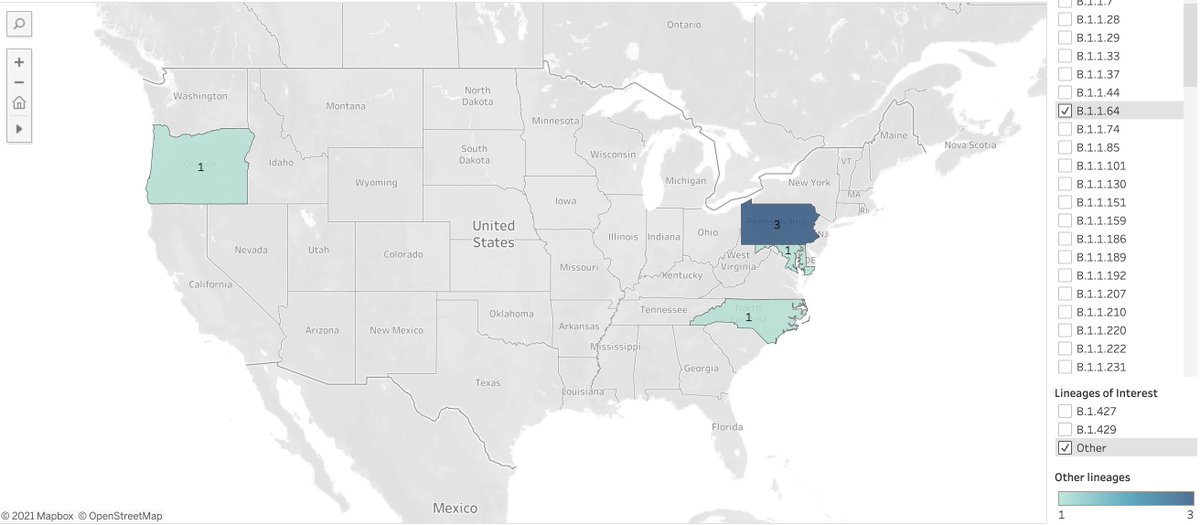
Random ex: B.1.1.64 in 4 states (note: I don't know anything about this variant).
public.tableau.com/profile/helix6…
- SGTF info up to Feb 15
- Seq info up to Jan 30
🧵 with more results
- New dashboard: you can check all lineages now
Random ex: B.1.1.64 in 4 states (note: I don't know anything about this variant).
public.tableau.com/profile/helix6…
- SGTF info up to Feb 15
- Seq info up to Jan 30
🧵 with more results
2/
Of the variants of concern, so far we only identified B.1.1.7 (666 times up to Jan 30). No B.1.351 and no P.1
We identified many B.1.429, a variant of interest. It represents about 20% of the sequences we do every day. But sampling is still biased for SGTF
Of the variants of concern, so far we only identified B.1.1.7 (666 times up to Jan 30). No B.1.351 and no P.1
We identified many B.1.429, a variant of interest. It represents about 20% of the sequences we do every day. But sampling is still biased for SGTF

3/
To assess fraction compared to non-SGTF sequenced, you can also get that info from the 2 files on Github with ALL of the data.
Check it out
github.com/myhelix/helix-…
- Info on >300,000 positive tests in last 2.5 months
- Info on 2,843 sequences (2/3 are random non-SGTF)
To assess fraction compared to non-SGTF sequenced, you can also get that info from the 2 files on Github with ALL of the data.
Check it out
github.com/myhelix/helix-…
- Info on >300,000 positive tests in last 2.5 months
- Info on 2,843 sequences (2/3 are random non-SGTF)
4/
Tip: if you want to quickly download the 2 files on your computer.
- Click on the file on home page (github.com/myhelix/helix-…).
- Then click on "Raw". see picture.
- Then on your Chrome toolbar: File/Save Page as
=> your turn to play.
Tip: if you want to quickly download the 2 files on your computer.
- Click on the file on home page (github.com/myhelix/helix-…).
- Then click on "Raw". see picture.
- Then on your Chrome toolbar: File/Save Page as
=> your turn to play.

5/
It is very rewarding to see many use this information.
Example: @scottleibrand analysis of what might happen in next few months
It is very rewarding to see many use this information.
Example: @scottleibrand analysis of what might happen in next few months
https://twitter.com/scottleibrand/status/1362330346763603969?s=20
6/
@trvrb also referenced @my_helix work in this great thread of what we might expect in next few months.
@trvrb also referenced @my_helix work in this great thread of what we might expect in next few months.
https://twitter.com/trvrb/status/1362438562449293312?s=20
7/
To finish, the usual snapshot of % of SGTF in Florida
Feb 14, 5-day avg was: 16% of positive tests are SGTF.
And experimental evidence support that >90% of these are B.1.1.7
Overall cases going down, vaccination is up, B117 is up. We keep tracking.
@ScottGottliebMD
To finish, the usual snapshot of % of SGTF in Florida
Feb 14, 5-day avg was: 16% of positive tests are SGTF.
And experimental evidence support that >90% of these are B.1.1.7
Overall cases going down, vaccination is up, B117 is up. We keep tracking.
@ScottGottliebMD

8/
Credit to many Helix teams. In particular @genesareclever and William Lee for leading the effort.
See our preprint for all involved.
medrxiv.org/content/10.110…
Credit to many Helix teams. In particular @genesareclever and William Lee for leading the effort.
See our preprint for all involved.
medrxiv.org/content/10.110…
9/
So we made it a little easier. There is now a page with the fraction of B.1.427 or B.1.429 compared to non-SGTF sequences.
public.tableau.com/profile/helix6…
- check US or by state.
- look at total non-SGTF seq per collection day
Jan 29, 18% of non-SGTF we sequenced were B.1.429
So we made it a little easier. There is now a page with the fraction of B.1.427 or B.1.429 compared to non-SGTF sequences.
public.tableau.com/profile/helix6…
- check US or by state.
- look at total non-SGTF seq per collection day
Jan 29, 18% of non-SGTF we sequenced were B.1.429

• • •
Missing some Tweet in this thread? You can try to
force a refresh










