
#SARSCoV2 selection analyses updates. We switched to running sliding windows analyses (blocks of 3 months) to deal with data volumes and get temporal trends. The current state of analyses is at observablehq.com/@spond/selecti…
This includes an at-a-glance view of selection profiles on the most recent time window 

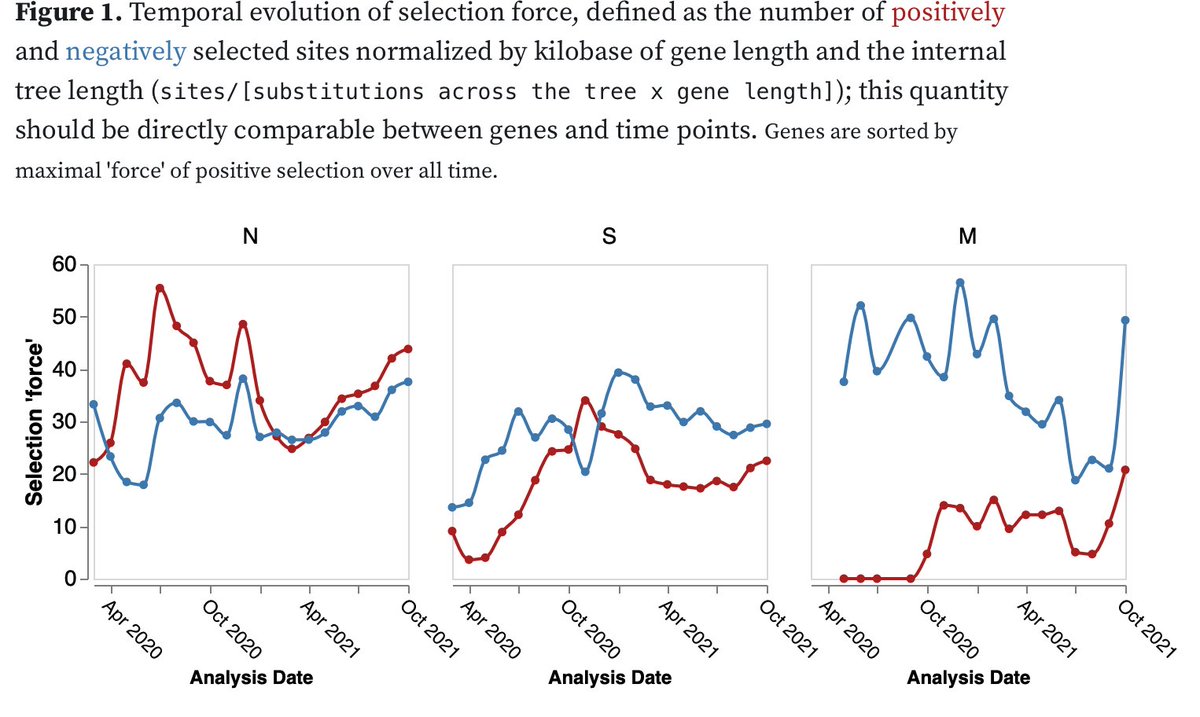
Profiles of selective "forces"
And evolutionary history of any subset of genomic sites (here the metasignature from cell.com/cell/pdf/S0092…)
observablehq.com/@spond/sars-co…
observablehq.com/@spond/sars-co…

• • •
Missing some Tweet in this thread? You can try to
force a refresh


