First @UKHSA Tech Briefing of the #Omicron era. A lot of scene setting, as it is very early days (I'd guess there's going to be a bit more info in next week's report, and loads more the week after that). But a few early things I think worth noting: 🧵
Annotated table of defining mutations is quite handy, and runs over six pages (this lineage has a lot of mutations). 

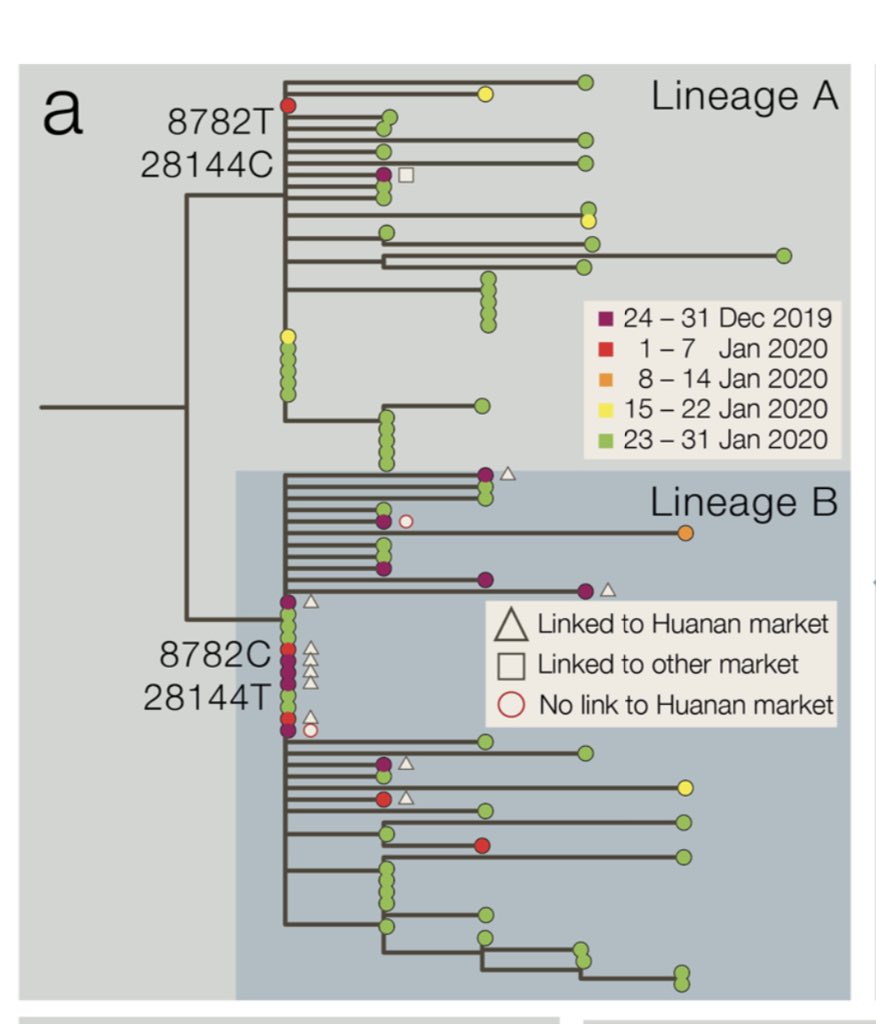
The 22 UK sequences in this report sit all over the global tree, suggesting multiple recent imports. This picture will change fast as we add more sequences.

There was no Omicron in the UK in the summer. Trust me.

One mutation in Omicron allows it to be picked up with a particular signature in the diagnostic PCR. This "SGTF" has been the leading indicator that Omicron is now dominant in South Africa, and has started going up in UK testing labs.
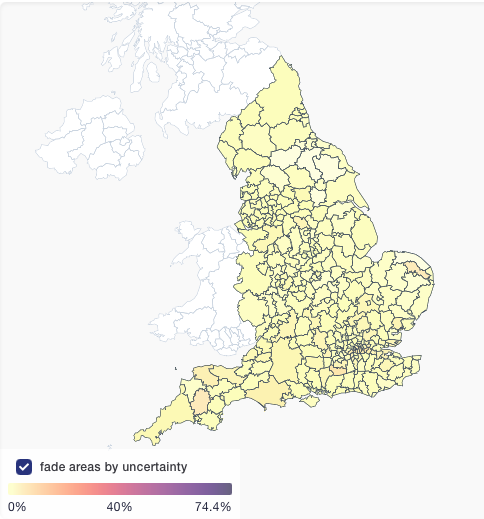
I had a quick look at the three biggest labs supplying @sangerinstitute sequencing, and you can see this leading indicator in the past couple days is going up fast, and the sequences are just about to catch up. (crisis mode means my..ahem..basic figures get into the report)

So my main take-away is that growth looks fast in a country with immunity mostly from vaccines, rather than previous infection (and with a big ongoing Delta wave). We'll know a lot more soon, but this is not reassuring.
• • •
Missing some Tweet in this thread? You can try to
force a refresh