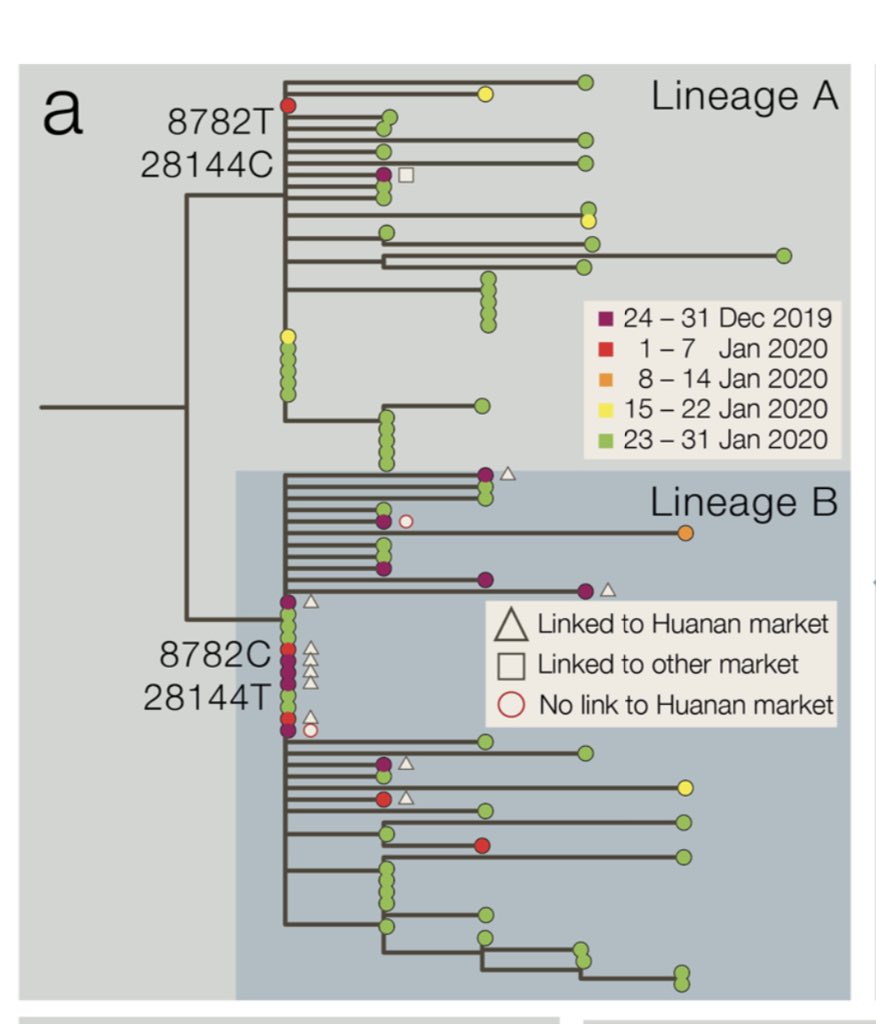
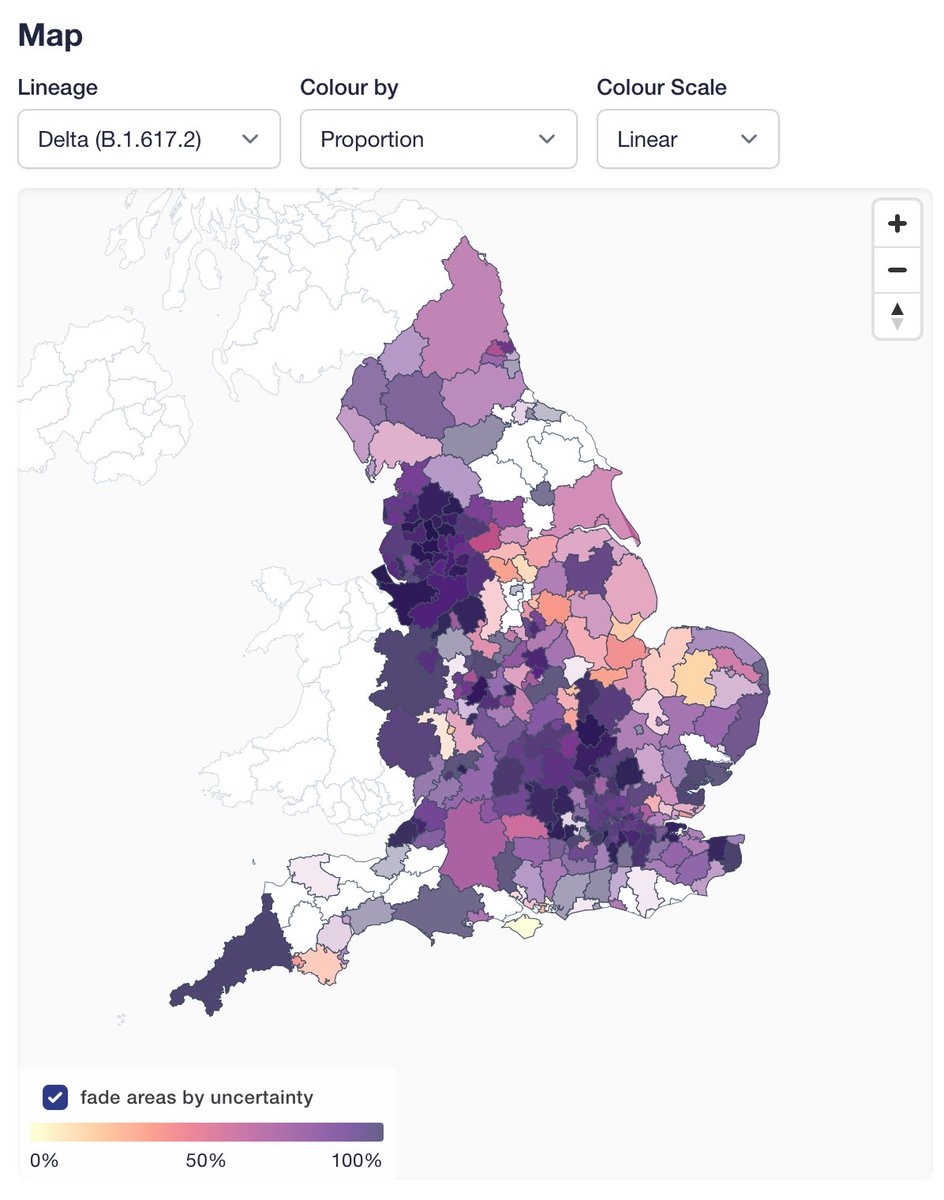
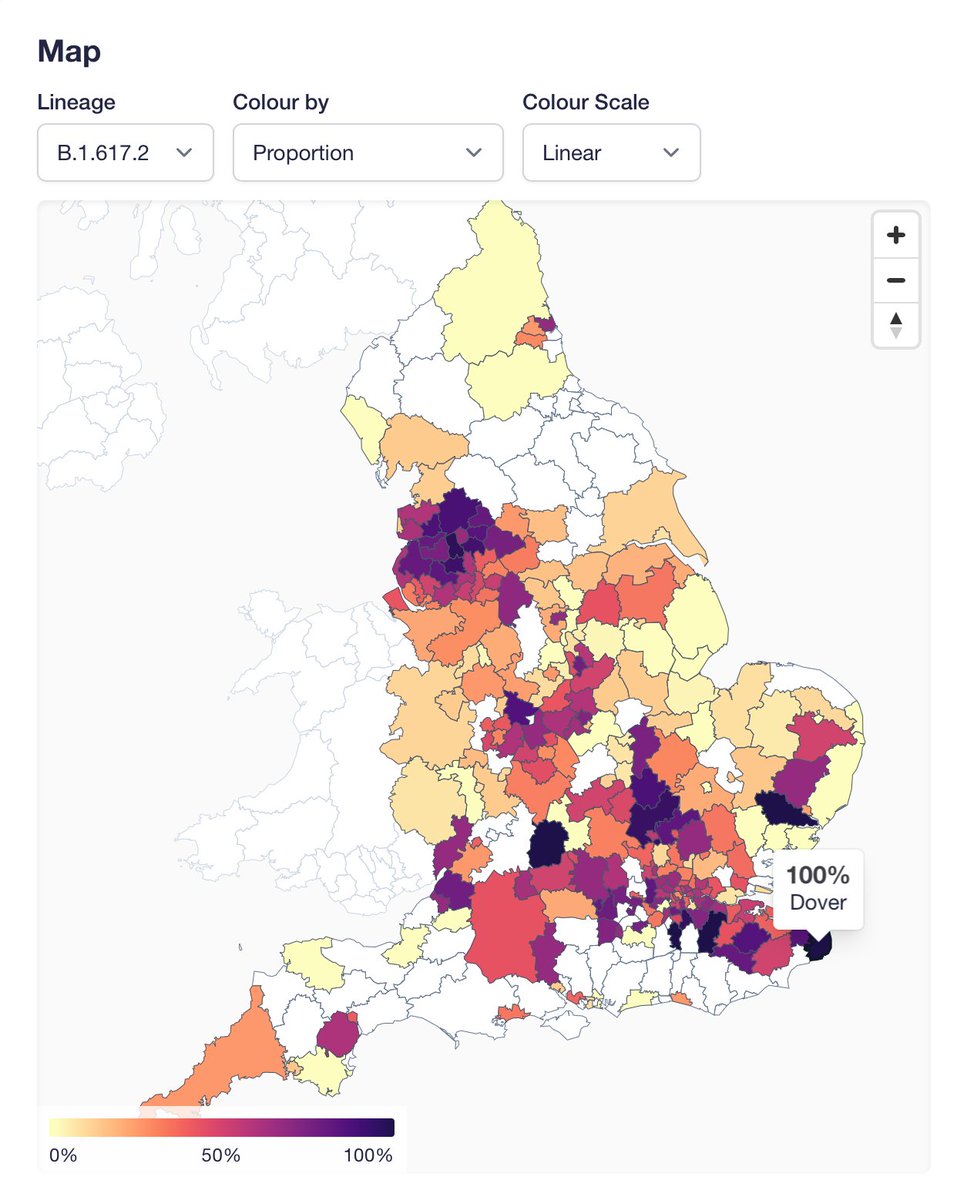
Proportion of AY.4.2 (now on covid19.sanger.ac.uk) has been steadily increasing in England, which is a pattern that is quite different from other AY lineages. Several of them rose when there was still Alpha to displace, but none has had a consistent advantage vs other Delta. 

And it has grown all over the place between mid-July (L) and now (R). AY.4 did a similar thing, but it was not displacing other Delta, and given that it hasn't spread through the world, likely just had some epidemiological luck.
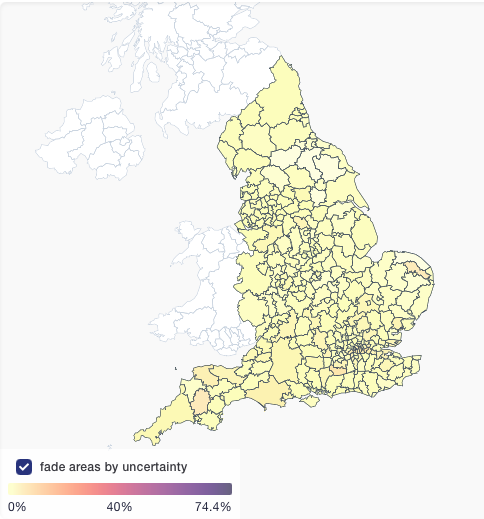

AY.4.2 hasn't yet spread much outside the UK, so could be a fluke (no biological advantage), but given that it's apparent advantage is modest, it needs to really get established somewhere to overcome stochasticity of small numbers.
That is, landing 20 Delta infections in a place with only Alpha almost guarantees that it will go on to complete replacement, whereas 20 AY.4.2 landing in an existing Delta epidemic may well fizzle.
10-15% increase in growth rate would be annoying but not catastrophic for the coming months. Likely the reason we're seeing this in the UK is a combination of high case rates (more mutations happening) and high sequencing rates (see changing patterns quickly).
What is perhaps more worrying is that it suggests the virus still has evolutionary paths to higher transmissibility open to it, and there are millions of Delta cases around the world without much sequencing coverage.
• • •
Missing some Tweet in this thread? You can try to
force a refresh