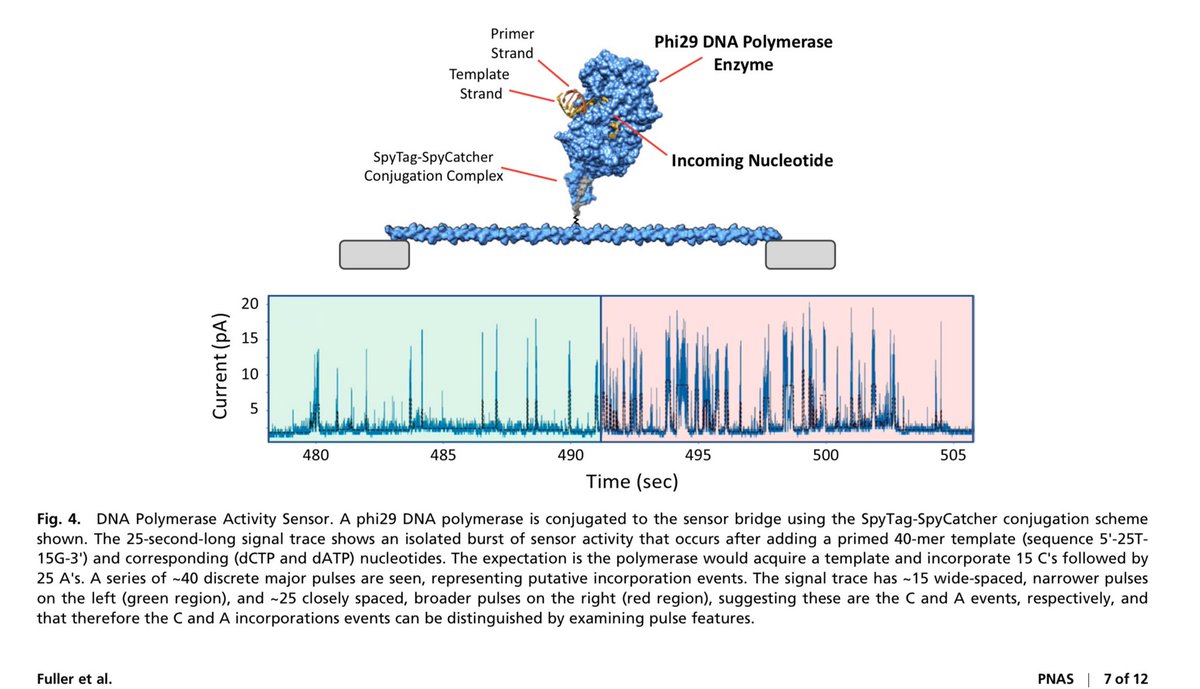
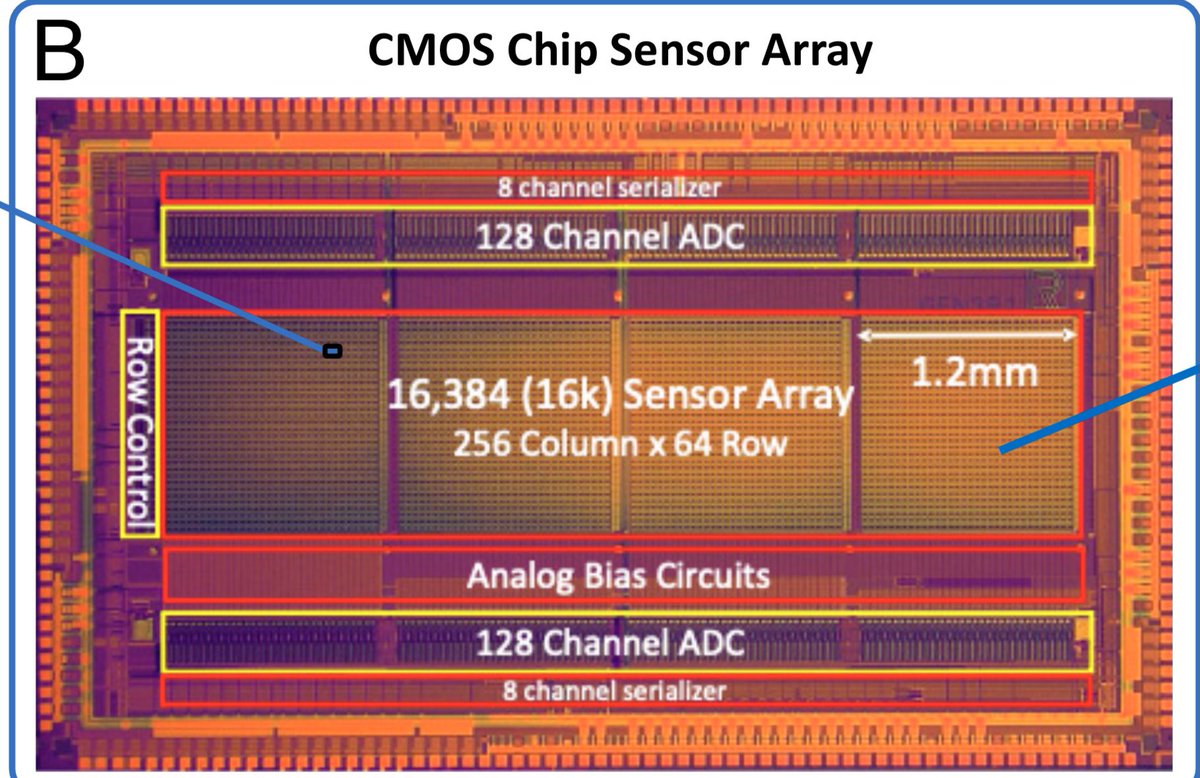
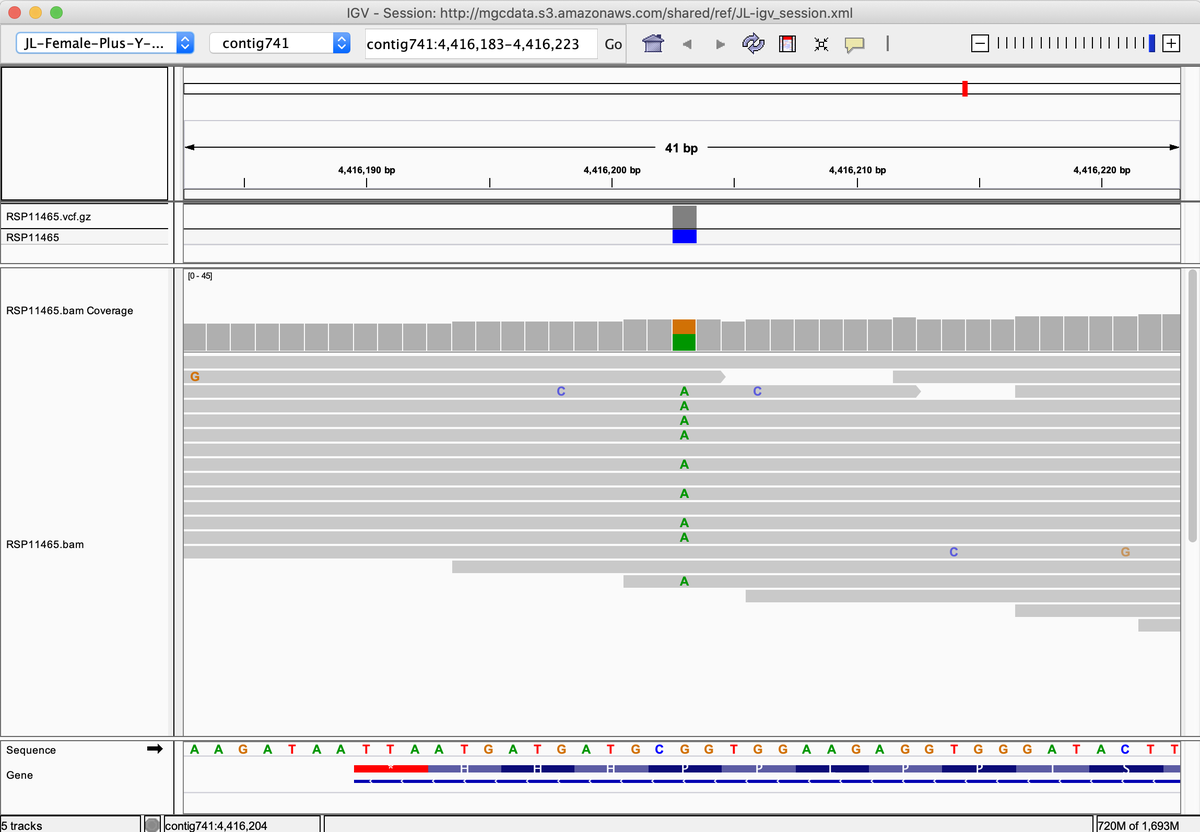
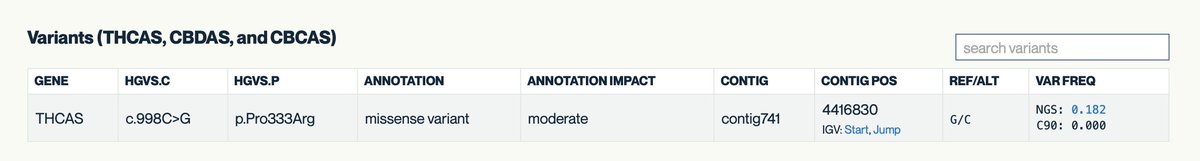
The Antartica metagenomic samples that @jbloom_lab nicely covered has some quirks.
NCBI claims its Illumina Data
Fastq files have headers that look like MGISeq data.
So I decided to take a look at their Adaptor sequences as each sequencer has their own unique flow cell primers
NCBI claims its Illumina Data
Fastq files have headers that look like MGISeq data.
So I decided to take a look at their Adaptor sequences as each sequencer has their own unique flow cell primers
https://twitter.com/stevenemassey/status/1491539897399791624
TrimGalore indeed confirms these are MGISEQ reads.
This paper has the MGISEQ adaptor sequences-
frontiersin.org/articles/10.33…
This paper has the MGISEQ adaptor sequences-
frontiersin.org/articles/10.33…
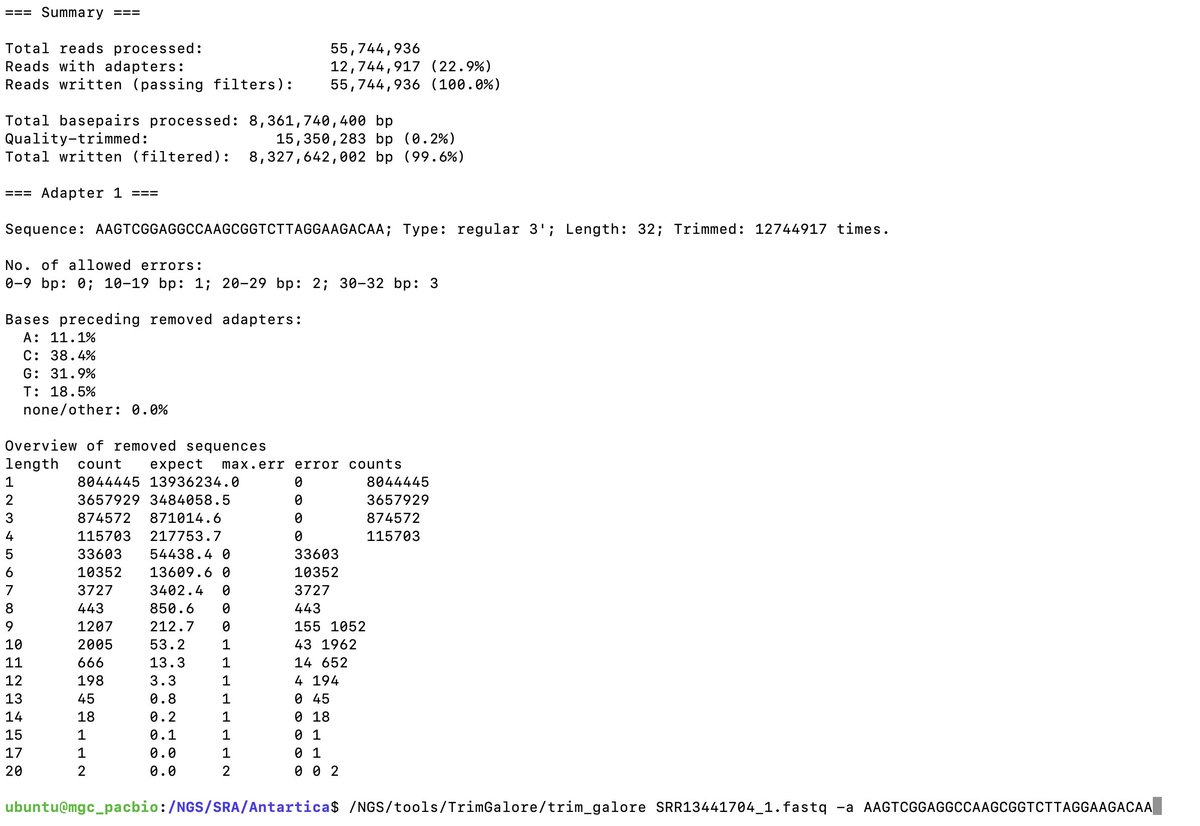
Why does this matter? The authors posit that this could be a result of the high index hopping problem seen on Illumina platforms.
MGISEQs documented index hopping rate is orders of magnitude lower than Illumina.
bmcgenomics.biomedcentral.com/articles/10.11…
MGISEQs documented index hopping rate is orders of magnitude lower than Illumina.
bmcgenomics.biomedcentral.com/articles/10.11…
This implies the contamination would have to occur prior to index ligation and the SARs construct would mostly likely have to be DNA not RNA for a DNA based metagenomic library to capture it. Has anyone looked for vector sequences in the data?
Why do we care about vector sequence? That implies human manipulation in dec 2019.
If index hopping is ruled out,
Then the contamination must be earlier and RNA molecules eventually must be turned into DNA for metagenomic libraries to capture them.
Someone had it as cDNA/Vector
If index hopping is ruled out,
Then the contamination must be earlier and RNA molecules eventually must be turned into DNA for metagenomic libraries to capture them.
Someone had it as cDNA/Vector
I have send the authors this thread. Their finding of cells line DNA in the metagenomic DNA is also suggestive of human manipulation prior to December 2019.
After using the MGISEQ adaptors for trimming we get more Forward reads to map but still 2X more reverses. The MGISEQ has more signal on the reverse reads as there is an additional polymerase replication event.
However the reads look very noisy on their 3' ends. Sign of dim DNBs
However the reads look very noisy on their 3' ends. Sign of dim DNBs
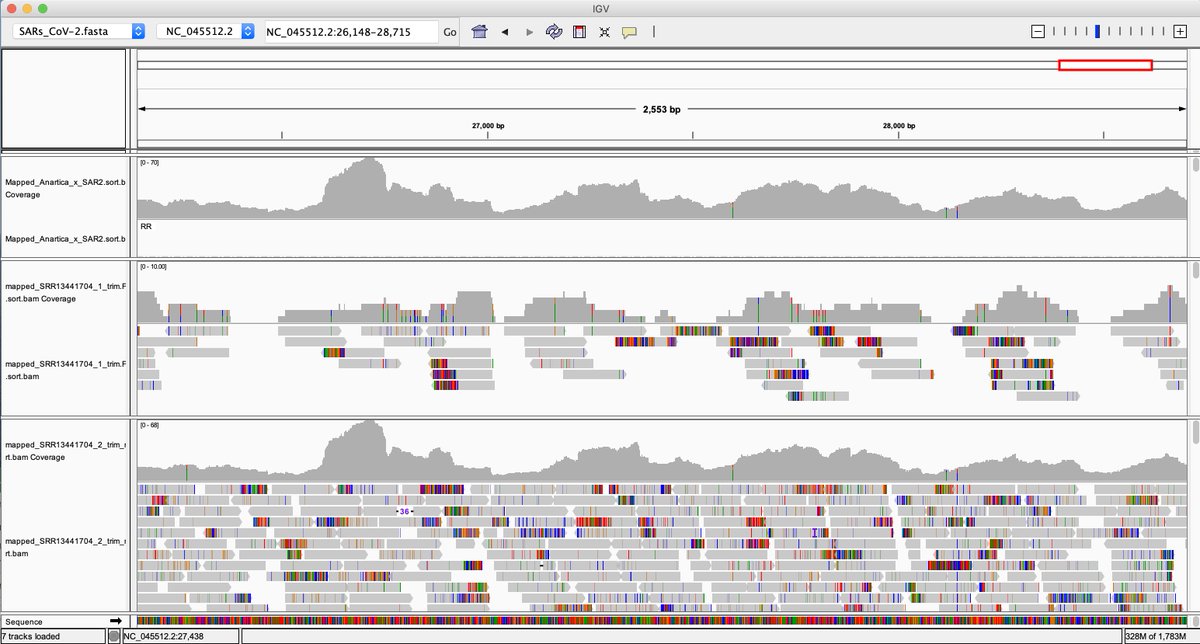
Dim DNBs usually have lower quality and are more prone to index hopping from neighboring bright DNBs.

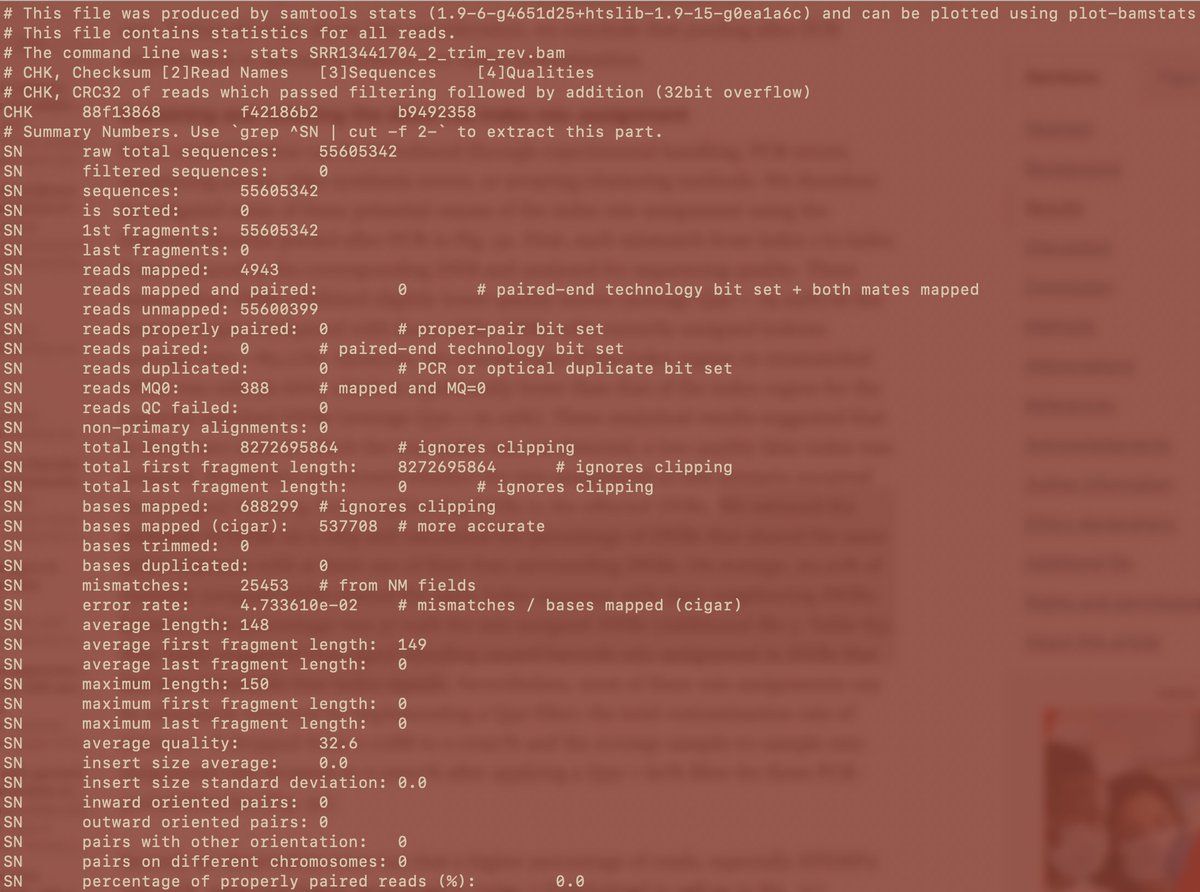
This is quality of the Reverse reads that mapped and all Reverse reads. There is a 10Q difference. Q20 reads have 1 error every 100bp. Q30 reads have 1 error every 1,000 bases. That 10 fold drop in quality means Index hopping may still be on the table. We 6K/55M reads mapping.

• • •
Missing some Tweet in this thread? You can try to
force a refresh