Carl Fuller et al are at it again.
This is wild. 16,384 CMOS chip capable of single molecule movies at each pixel.
Each pixel has a protein nanowire bridging it. These are assembled with electrophoresis.
Once wired, click chemistry binds one molecule.
pnas.org/content/pnas/1…
This is wild. 16,384 CMOS chip capable of single molecule movies at each pixel.
Each pixel has a protein nanowire bridging it. These are assembled with electrophoresis.
Once wired, click chemistry binds one molecule.
pnas.org/content/pnas/1…
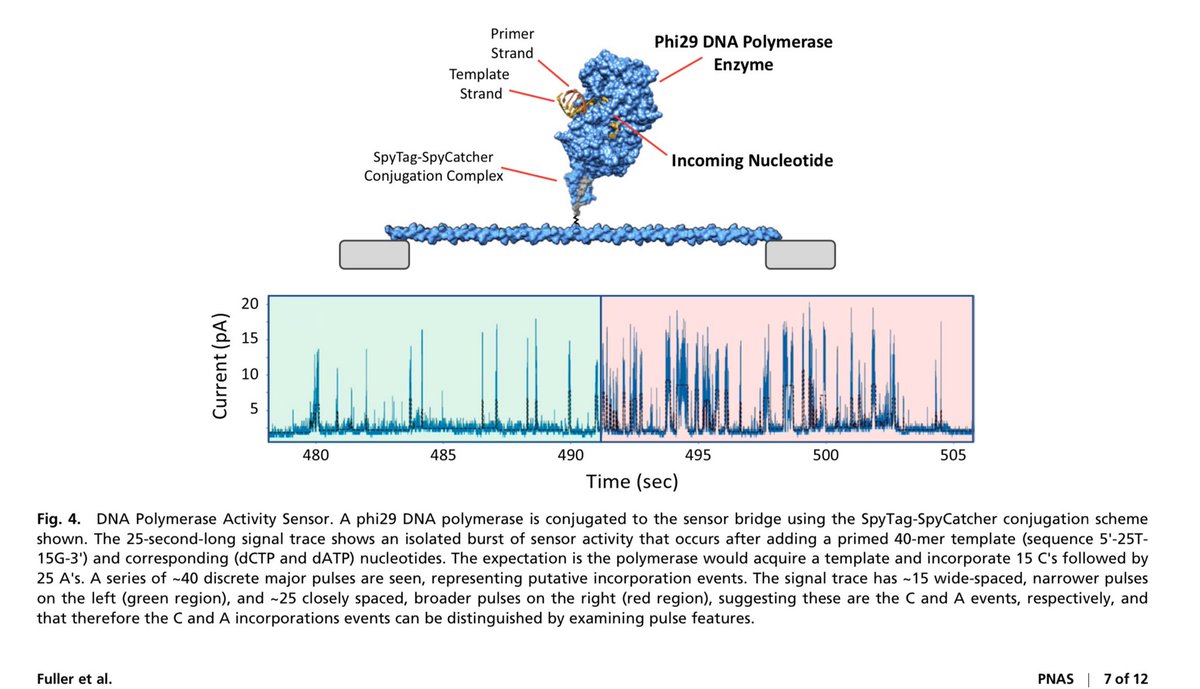
20nM gap for the nanowire peptide bridge.
1000Hz read outs. 0-400pAmp sensors.
1000Hz read outs. 0-400pAmp sensors.
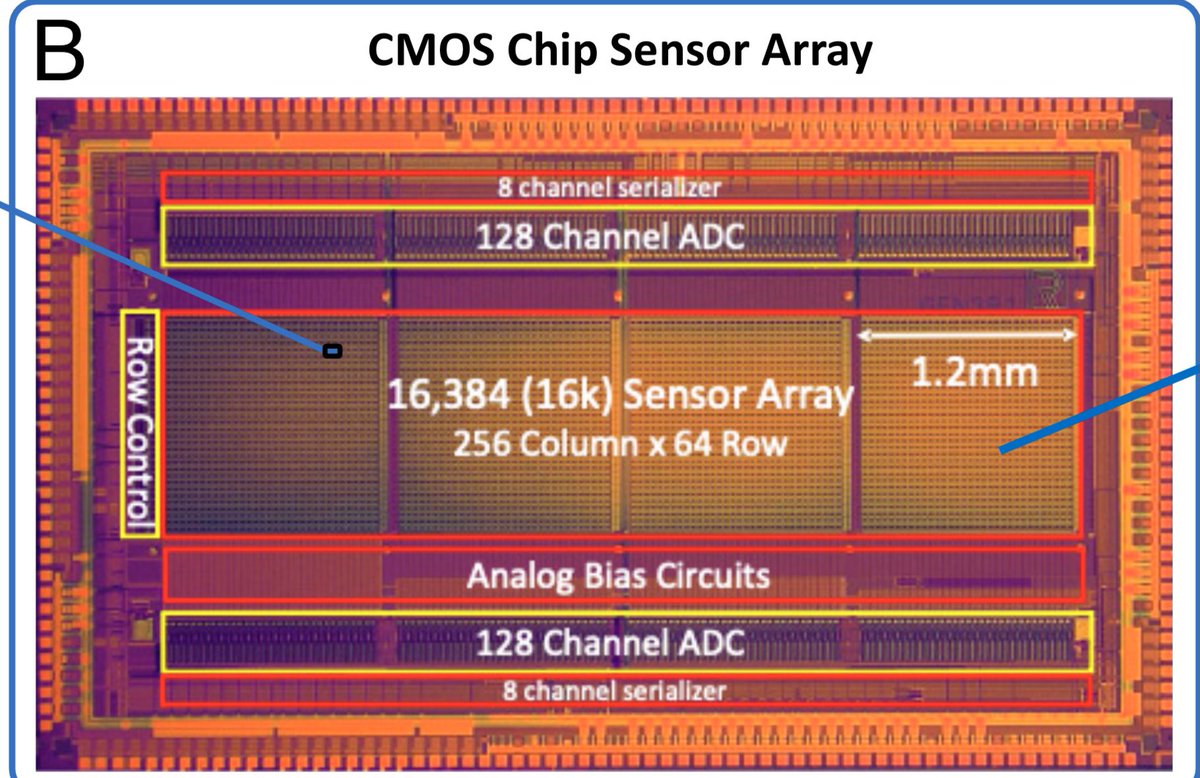
Chip features

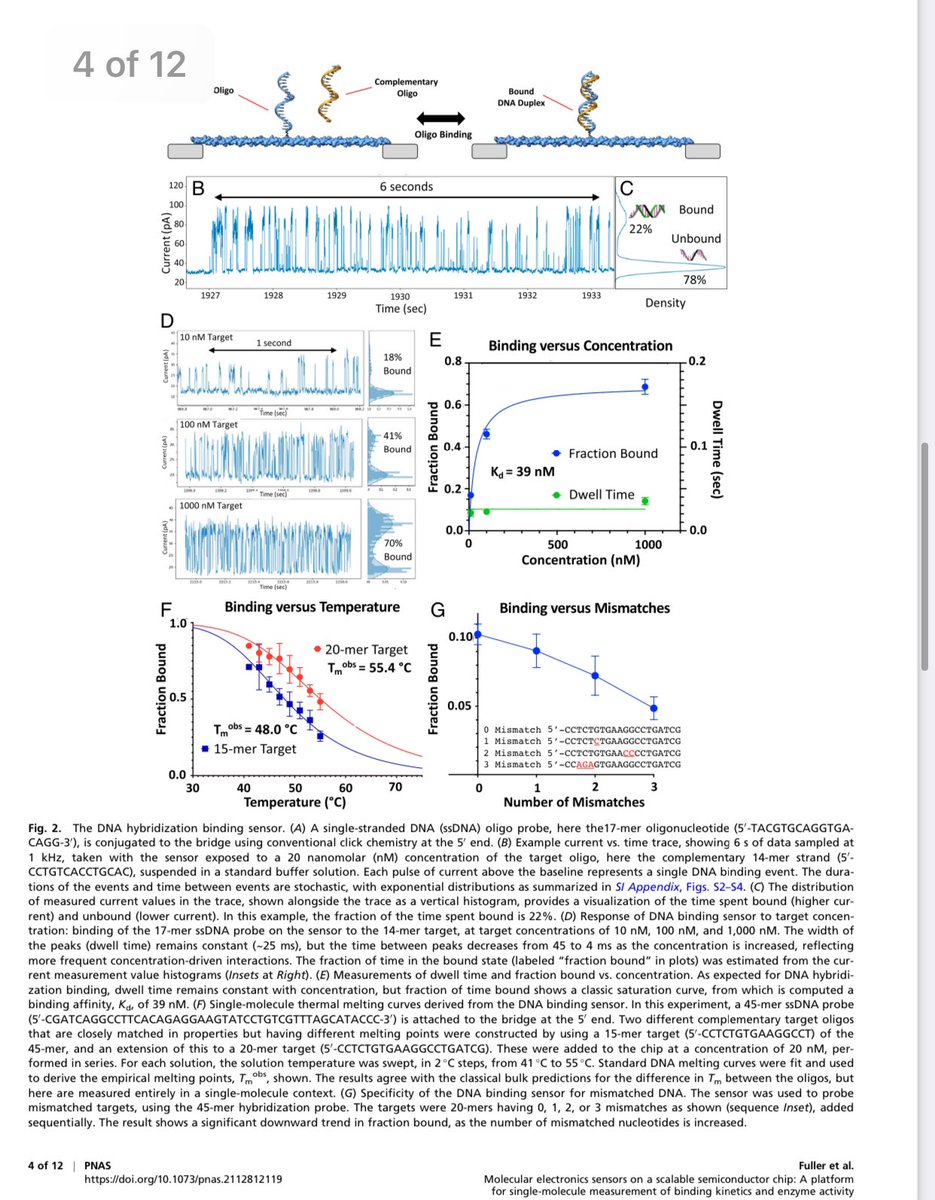
Simple Oligo binding assays are information rich.
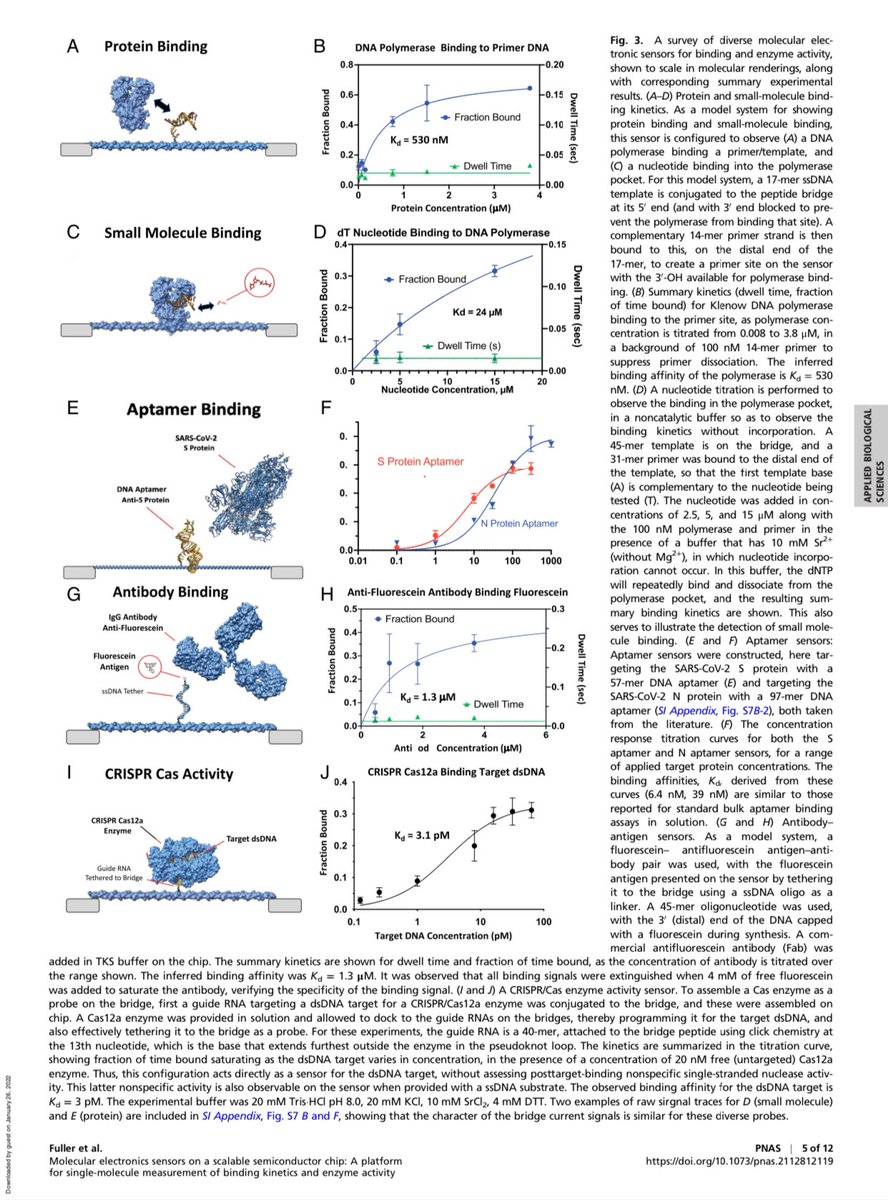
The diversity of applications is impressive.
• • •
Missing some Tweet in this thread? You can try to
force a refresh