New🔥 #DataScience #Bioinformatics resource: 850,000‼️ #scRNAseq cells from 226 samples across 10 cancer types draw a map of the tumor microenvironment, in particular fibroblasts.
Let’s see👇what are the main contributions of this work & what this means for #cancer #Genomics🧵
Let’s see👇what are the main contributions of this work & what this means for #cancer #Genomics🧵
But first, some background.
Cancers are (unfortunately) complex ecosystems,consisting of various types of cells.
Malignant cells represent only a fraction of the tumor. The rest is made of the tumor microenvironment/TME (fibroblasts + immune cells), with complicated dual roles.
Cancers are (unfortunately) complex ecosystems,consisting of various types of cells.
Malignant cells represent only a fraction of the tumor. The rest is made of the tumor microenvironment/TME (fibroblasts + immune cells), with complicated dual roles.

Understanding the essence of this duality is key in understanding why most cancer therapies fail.
TME cells are plastic & can easily change states.
The same TME cells can either promote or suppress tumor development, depending on very subtle factors totally not well understood.
TME cells are plastic & can easily change states.
The same TME cells can either promote or suppress tumor development, depending on very subtle factors totally not well understood.
The immune system, due to its remarkable capacity to be stimulated and fight 🤺tumor expansion, has been the object of several detailed #singlecell profiling studies, both zooming in on specific cancer types, as well as across cancers (see example👇)
https://twitter.com/simocristea/status/1577313316120059904?s=20&t=X-2KcDDHsH3CKlfBygwvjg
Impressive recent works have dissected immune mechanisms in great detail,using novel innovative techniques. Here’s one specific example👇of the complex dual role that immune cells can play in the life of a tumor (both immuno-suppressive & immuno-promoting)
https://twitter.com/simocristea/status/1583499183561900033?s=20&t=X-2KcDDHsH3CKlfBygwvjg
On the contrary, fibroblasts have been the TME component less studied, despite their known roles in influencing disease outcome by e.g. interacting with T cells or promoting angiogenesis.
For ex, cancer associated fibroblasts (CAF) have multiple documented tumor promoting roles.
For ex, cancer associated fibroblasts (CAF) have multiple documented tumor promoting roles.
Back to the current paper:
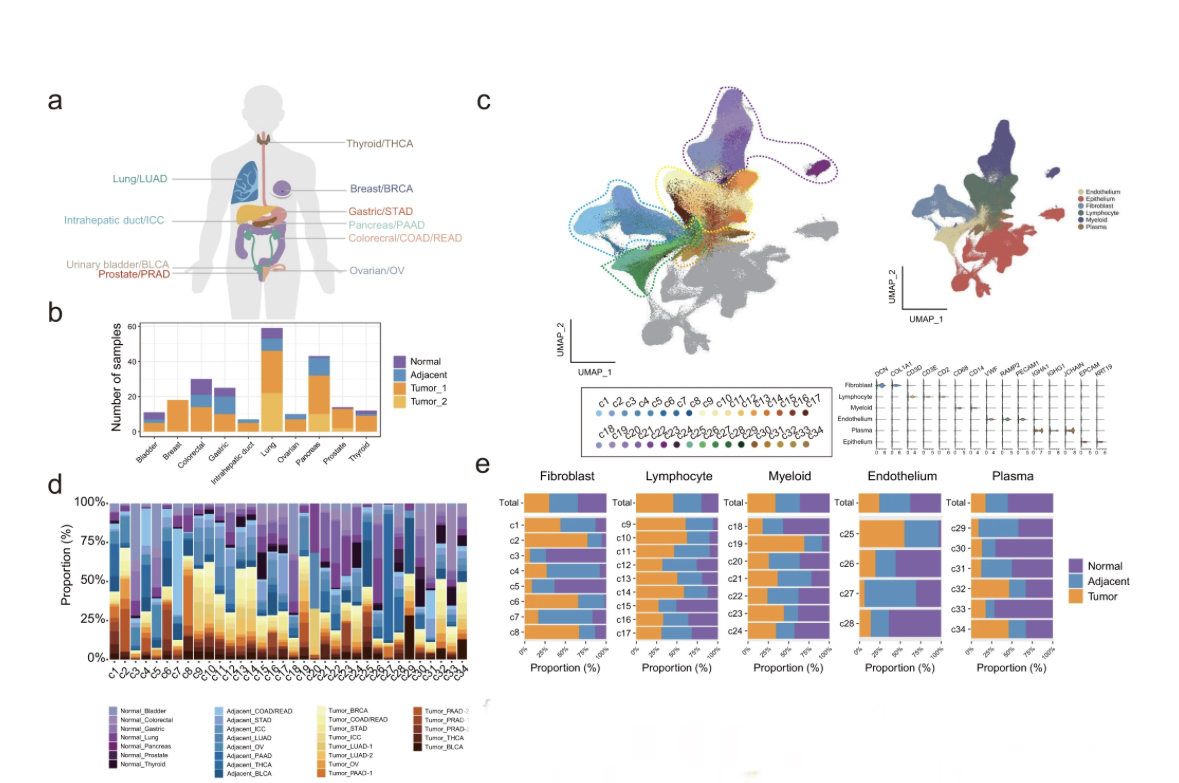
A compilation of 148 primary tumor, 53 adjacent, and 25 normal samples from 164 donors enrolled in 12 studies (part newly sequenced, part public).
This corresponds to almost 900K unsorted #scRNAseq cells after QC, belonging to various cell types.
A compilation of 148 primary tumor, 53 adjacent, and 25 normal samples from 164 donors enrolled in 12 studies (part newly sequenced, part public).
This corresponds to almost 900K unsorted #scRNAseq cells after QC, belonging to various cell types.

This data was batch-corrected up-front using a Seurat wrapper for the fastMNN algorithm in the batchelor R package @MarioniLab. This algorithm estimates batches by correcting average mutual nearest neighbor pairs found on a low-dimensional projection between reference and target.
Now, I would have liked to see the non-corrected data as well, to better understand what this correction is doing here. With patient samples, part of the “batch” can also be genuine biological variation (see👇). Unfortunately,this analysis is not provided.
https://twitter.com/simocristea/status/1578441680939732993?s=20&t=X-2KcDDHsH3CKlfBygwvjg
The major expected cell types were identified in the TME, including many epithelial cells.
I was surprised by the large epithelial fraction (~50% of data), since in my experience, @10xGenomics #scRNAseq kits preferentially enrich for (most) leukocytes & deplete epithelium/stroma
I was surprised by the large epithelial fraction (~50% of data), since in my experience, @10xGenomics #scRNAseq kits preferentially enrich for (most) leukocytes & deplete epithelium/stroma

This is mainly because leukocytes don’t mind being by themselves in a droplet, since they are used to hanging out alone, while epithelium/stroma like to hang out closer to other similar cells.
By analyzing the epithelial cells separately, a bias in state (malignant/normal) and tumor type was quite visible (even though not quantified).
In my experience, this is completely expected, as most of tumor heterogeneity comes from the characteristics of the epithelium.
In my experience, this is completely expected, as most of tumor heterogeneity comes from the characteristics of the epithelium.

Unlike epithelium, TME did not cluster by cancer type. Again to be expected, as:
1.TME transcriptional profiles are quite conserved across tumors
2.the different cell types in TME are indeed quite different
Good to see this confirmed here, across many patients & cancer tissues.
1.TME transcriptional profiles are quite conserved across tumors
2.the different cell types in TME are indeed quite different
Good to see this confirmed here, across many patients & cancer tissues.

Cell-cell interactome analysis showed how all main cell types identified in the TME had lots of crosstalk.
In particular, fibroblasts were the most prolific interactor within the TME, regardless of tissue type or malignancy status (cancer or adjacent normal)
In particular, fibroblasts were the most prolific interactor within the TME, regardless of tissue type or malignancy status (cancer or adjacent normal)

Now zooming into fibroblasts:
Canonical markers allowed classifying the three most important subtypes as cancer-associated myofibroblasts (CAFmyo), inflammatory CAFs (CAFinfla), and adipogenic CAFs (CAFadi).
Other components were present, but less specific or prominent.
Canonical markers allowed classifying the three most important subtypes as cancer-associated myofibroblasts (CAFmyo), inflammatory CAFs (CAFinfla), and adipogenic CAFs (CAFadi).
Other components were present, but less specific or prominent.
SCENIC regulatory analysis indicated that the activation of the CAF subtypes can be different.
Evolutionary trajectories deliniated 3 states distinctly evolving from normal fibroblasts (NFs) to CAFs: state1 (NFs dominant), state2 (CAFmyo dominant) & state3 (CAFadi/CAFinfla dom)
Evolutionary trajectories deliniated 3 states distinctly evolving from normal fibroblasts (NFs) to CAFs: state1 (NFs dominant), state2 (CAFmyo dominant) & state3 (CAFadi/CAFinfla dom)

CAFs are important to study because they can facilitate immunosurveillance escape.
Zoomed-in interactome analysis showed how CAFs do indeed interact with multiple types of identified immune cells (both innate, s.a. myeloid; & adaptive, s.a. T or B cells) in many complex ways.
Zoomed-in interactome analysis showed how CAFs do indeed interact with multiple types of identified immune cells (both innate, s.a. myeloid; & adaptive, s.a. T or B cells) in many complex ways.

Further zooming in to the 3 CAF states identified, if was found that
CAF differentiation may promote the stratification of patients with immunotherapy.
CAFstate3, which was at the most dedifferential state, predicted a worse outcome of immunotherapy in some cancer types.
CAF differentiation may promote the stratification of patients with immunotherapy.
CAFstate3, which was at the most dedifferential state, predicted a worse outcome of immunotherapy in some cancer types.

If further validated, this classification could contribute to stratification for both prognostic & therapeutic immunotherapy.
I very much like the last figure of the paper, in which the authors summarize their study.
CAFs are the bad guys here, and it’s these cells that we want to characterize well.
They can originate from four sources, but primarily they come from normal fibroblasts.
CAFs are the bad guys here, and it’s these cells that we want to characterize well.
They can originate from four sources, but primarily they come from normal fibroblasts.

Their activation trajectory is divided into 3 states (discussed above) associated with both immunomodulation in selected cancer types (via response to checkpoint inhibitors) & also with angiogenesis (via interactions with SPP1+ macrophages).

Finally, here is the link to the paper @NatureComms
nature.com/articles/s4146…
The data compiled here is a valuable resource for #cancer #genomics analyses.
It can also be quite easily further mined & visualized *interactively* via this nice portal:
gist-fgl.github.io/sc-caf-atlas/
nature.com/articles/s4146…
The data compiled here is a valuable resource for #cancer #genomics analyses.
It can also be quite easily further mined & visualized *interactively* via this nice portal:
gist-fgl.github.io/sc-caf-atlas/
• • •
Missing some Tweet in this thread? You can try to
force a refresh