After 4 years, it's rather nice to finally present our work on genetic's model trait, height, in >1.4M WES/WGS samples led by @doc_locke, @Mar_ferreira17, & @gabecasis where we found 207 genes [amongst many other results].
A thread of findings below⬇️
medrxiv.org/content/10.648…
A thread of findings below⬇️
medrxiv.org/content/10.648…

After conditioning on common variants, we found 207 genes (P<1.75e-9) via @marchini & @covariani's gene-P method to combine burden, SBAT, SKAT-O, & ACAT-V gene-based tests into 1 p-value. Burden tests find the majority of genes, but 28 (14%) are found only via SKAT-O/ACAT-V.
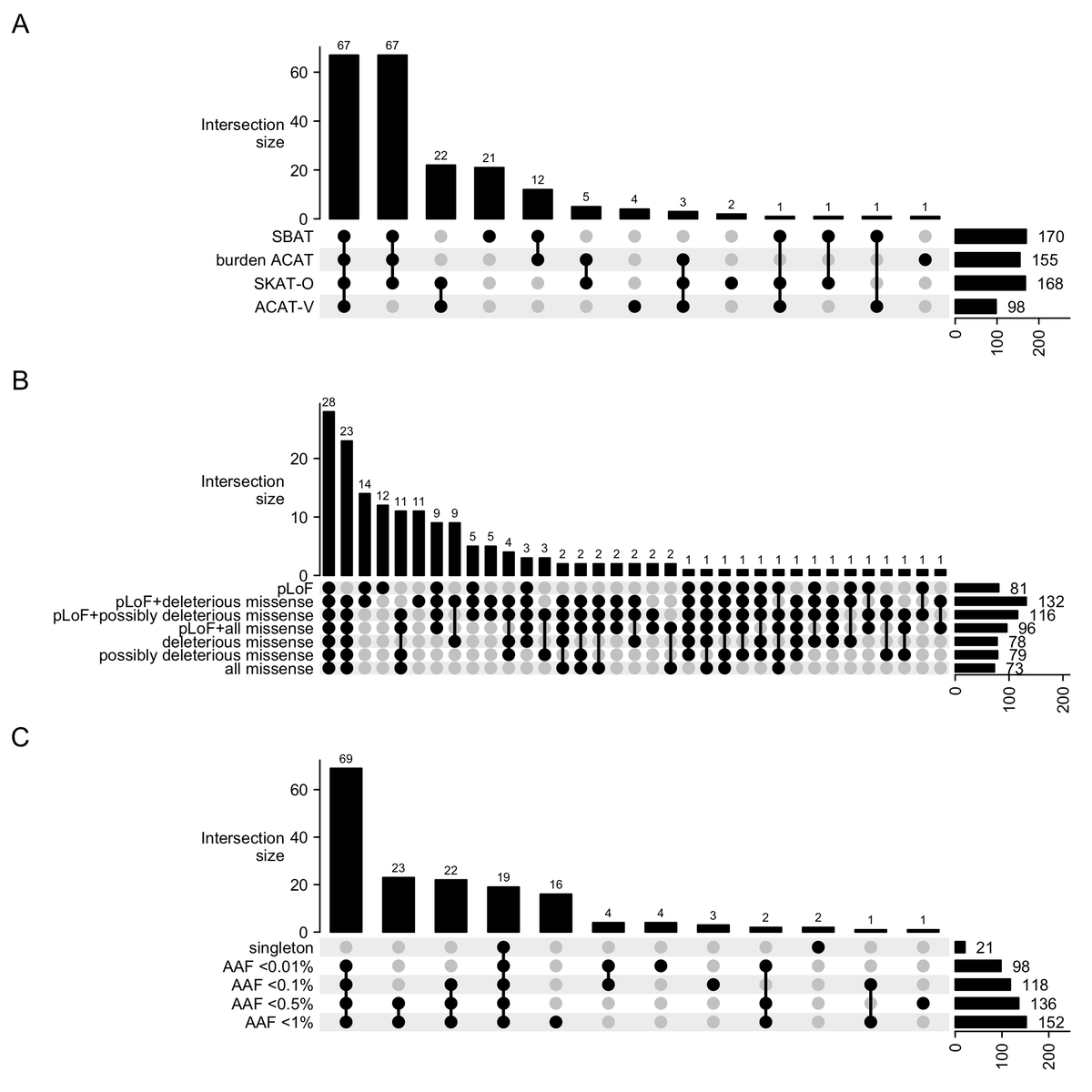
With burden tests, we observed the classic tradeoff between adding increasingly common variants to boost statistical power (via more allele counts) at the cost of weaker effect sizes.
Ex: We found 17 genes via singleton pLoFs (|β|=9cm) but 76 genes from <1% pLoFs (|β|=4cm).
Ex: We found 17 genes via singleton pLoFs (|β|=9cm) but 76 genes from <1% pLoFs (|β|=4cm).

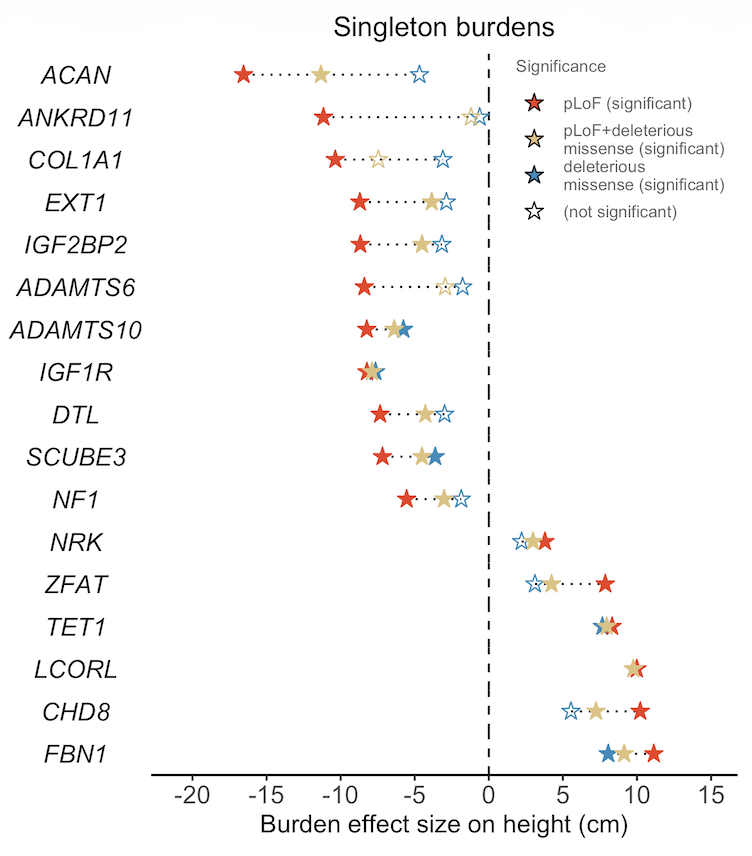
The most surprising result for me (obvious in hindsight🤦♂️) was finding well-known large effect, constrained, developmental genes like CHD8 (#ASD) or ANKRD11 (neurodevelopmental delay) by studying height - they do a lot more than just affect ASD or NDD!
Often ignored by papers are individual rare variants. After conditioning on GWAS loci, we found 107 rare nonsynonymous variants (P<1.75e-9) including FGFR3 & PTPN11 GoF missense variants with effects larger than singleton pLoF burdens (causing Acondroplasia & Noonan syndrome).
I found the analysis of individual rare variants to be quite insightful (shoutout to @doc_locke for suggesting it). B/c some genes' have large variance in per-variant βs, the burden β can be misleading. KMT2B burden 20x smaller than its largest missense variant (0.4cm v 7.7cm)
Inspired by one of my favorite papers by @HHeyne & @dalygene on recessive associations in FinnGen, I sadly only found 1 recessive association missed by the additive test - CFTR's delta508 mutation (recessive P=5e-10; additive P=0.14).
rdcu.be/fqfiK
rdcu.be/fqfiK
Lastly, my partner-in-crime, Liron Ganel, found an AMR-enriched missense variant in HHIP that lowers height by -4cm. This was even more interesting b/c it acts opposite to HHIP singleton pLoFs that increased height by +10cm.
• • •
Missing some Tweet in this thread? You can try to
force a refresh