A TWEETORIAL on our new CRISPR-sciATAC preprint. Our lab has been working on combining pooled CRISPR screens with single-cell chromatin accessibility for nearly 4 years so very happy to see this work finally out there in the world.
biorxiv.org/content/10.110…
biorxiv.org/content/10.110…
The work was led by 2 talented postdocs in our lab (@noaliscovitch and @antonino_nyc) with support from many other folks in our lab and in @NYGCTech lab. Extra shout-out to @noaliscovitch for getting the manuscript over the final hump in what has been a challenging year.
When we started, we were impressed by the diverse methods (e.g. ECCITE-seq, Perturb-seq, CROP-seq) for combining pooled CRISPR screens with single-cell RNA-seq and wondered if we could do something similar with chromatin accessibility (ATAC-seq).
Building off the pioneering single-cell combinatorial-indexing (sci)ATAC-seq from @cusanovich & @JShendure, we developed a method to add CRISPR guide RNA reverse transcription to the sciATAC workflow: CRISPR-sciATAC. 

This took a lot of trial-and-error to find a robust protocol for tagmentation followed by reverse-transcription in-situ (i.e. in nuclei). We identified a few key ingredients to doing this, including glyoxal fixation and a robust reverse transcriptase (RevertAid).

A major advantage here was an easy-to-purify transposase (TnY) from @psmibert @NYGCTech. This work would’ve been impossible using commercial transposases ($$$) and so TnY was really a breakthrough for us. Thanks Peter & @sumajaini for sharing this great tool!

With TnY and in situ RT, we mixed human and mouse cells to show that we could get good separation with human and mouse cells, a low doublet rate, and expected nucleosomal banding patterns.


We next targeted 20 chromatin modifiers that are highly mutated across cancers in human cells and performed CRISPR-sciATAC.
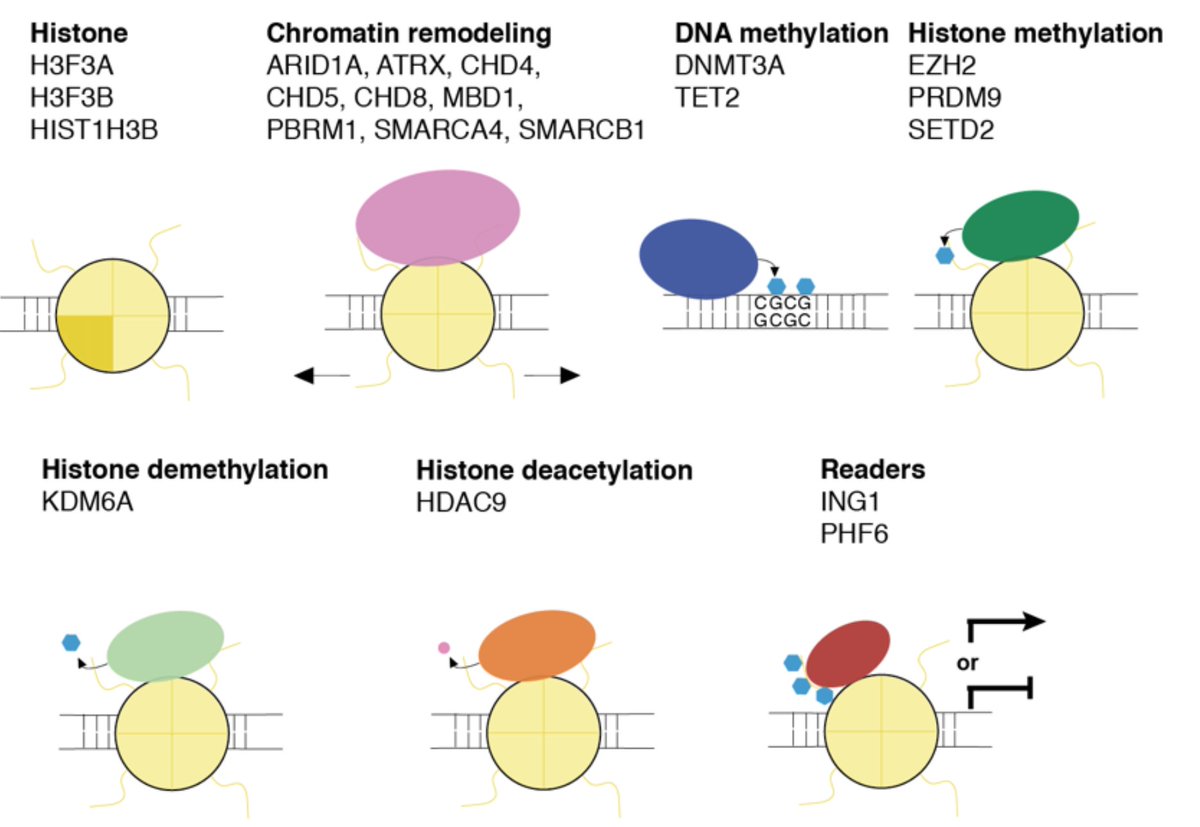

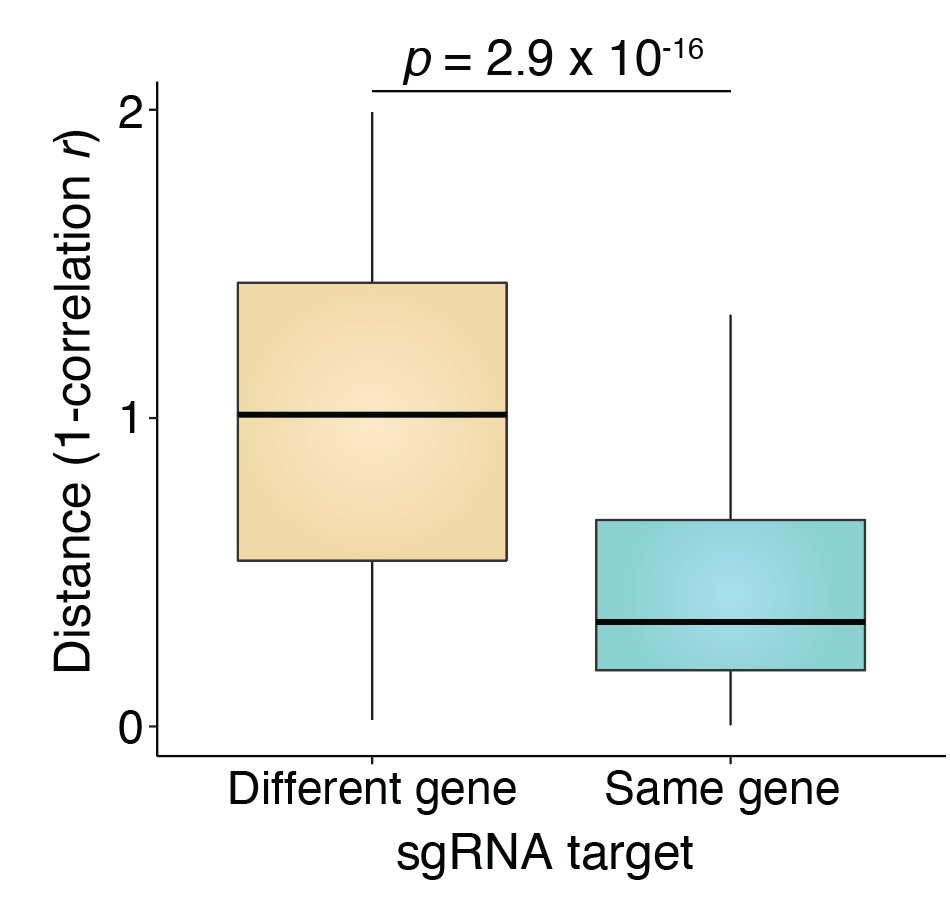
Here, we’re looking at chromatin accessibility overlapping specific histone/DNA modifications. Each row is a single Cas9 guide RNA — you can see there is a high degree of consistency between different guide RNAs that target the same gene.

One of the strongest effects can be seen with the Polycomb repressor EZH2. Here, we can separate single cells based on EZH2 perturbation status. Even a very small number of EZH2-perturbed cells has high correlation with the bulk.


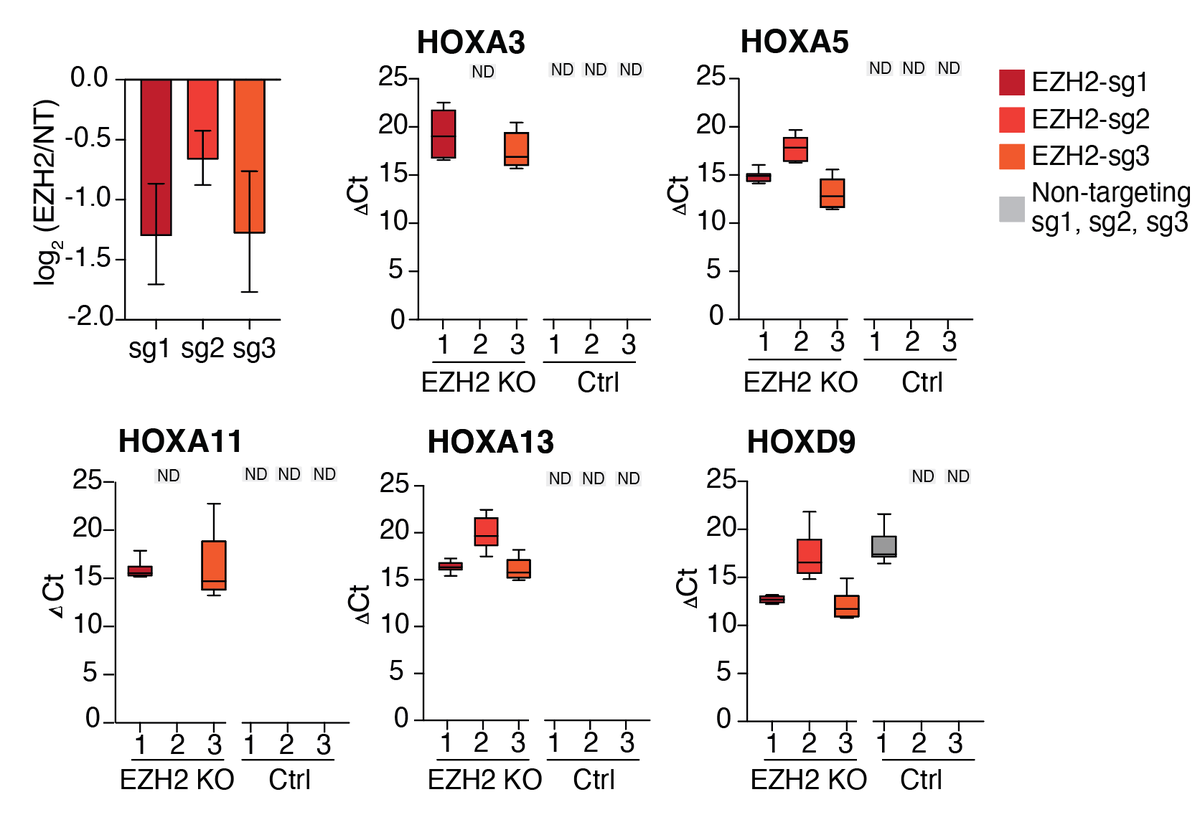
Looking at specific regions of the genome, we noticed that some of the HOX loci, like HOXA, had particularly large changes in accessibility upon loss of EZH2.
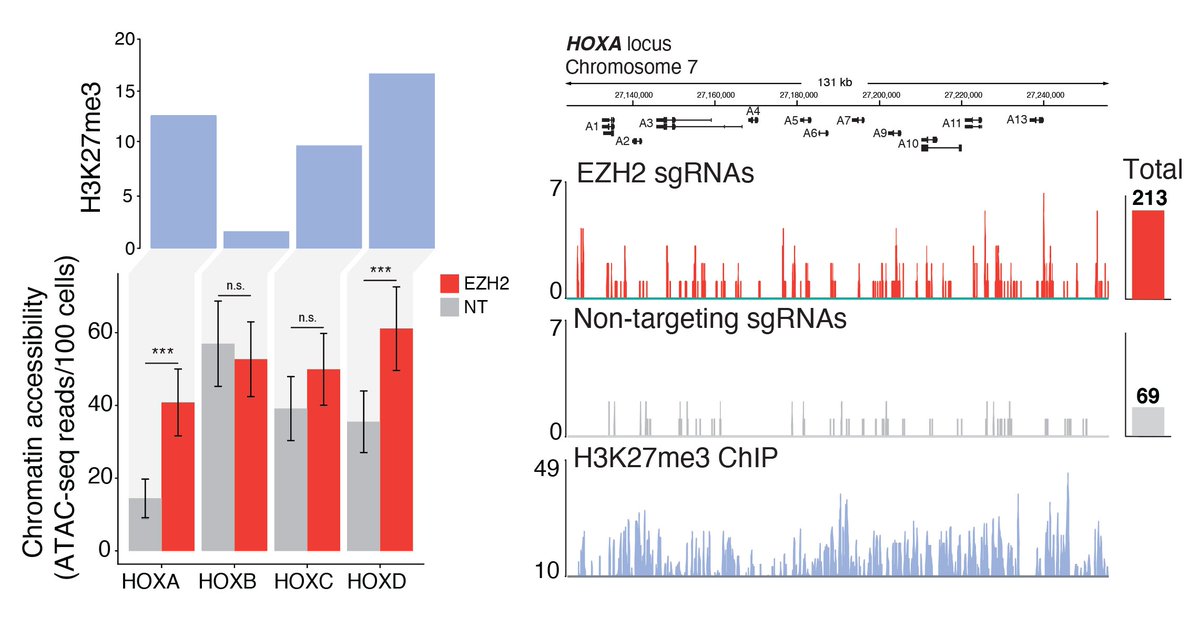
We wondered if the increase in open chromatin would result in a change in gene expression, so we took the same 3 CRISPR guide RNAs and measured the expression of 5 different HOX genes (along with EZH2) after EZH2 knock-out. These previously silenced genes are now expressed!
One exciting aspect of single-cell ATAC-seq is that we can examine co-regulatory relationships between different TFBS. For example, in EZH2 KO cells, we find that correlations in accessibility between CREBBP and other TF binding sites are no longer present.

Perhaps unsurprisingly, the disrupted co-regulatory TFBSs are enriched for pathways involved in cancer-related transcription.

After this small-scale experiment, we next decide to scale UP (!) and target the subunits of every chromatin remodeling complex in the EpiFactors database. ✂️💪🙌

Using nearly 17,000 CRISPR-perturbed single cells, we found that loss of subunits within chromatin remodeling complexes results in broadly-speaking, either increases in accessibility (eg. NuRF, CoREST) or decreases (eg. CREST, pBAF). FYI: Finer differences w/ individual subunits.

With dimensionality reduction on accessibility in transcription factor binding sites (TFBS), we found that SWI/SNF subunits clustered distinctly, likely due to disruption of many TFBS (in red in the bar plot).

Looking at individual gene knock-outs, you can see that some have broad effects (many TFBS), whereas loss of other subunits changes chromatin accessibility at just one TFBS. This is a fascinating diversity of effects in the #PerturbationAtlas ! (Can we get that trending? 😉)

We also looked to see if loss of certain chromatin remodeler subunits resulted in significant changes in promoters or enhancers or both. For example, ARID1A loss changes accessibility in enhancers (decreased accessibility) but not in promoters. How does this specificity arise?!?


Not satisfied with just measuring changes at the level of TFBS and promoters/enhancers, Noa built a pipeline to measure shifts in nucleosome position upon loss of chromatin remodelers/modifiers.

For TFBS with symmetric nucleosome positions (previously described by @anshulkundaje & others), we examined shifts in nucleosome spacing. In most cases, we found that CRISPR perturbations led to expansion of nucleosome spacing but there is quite a bit of diversity!
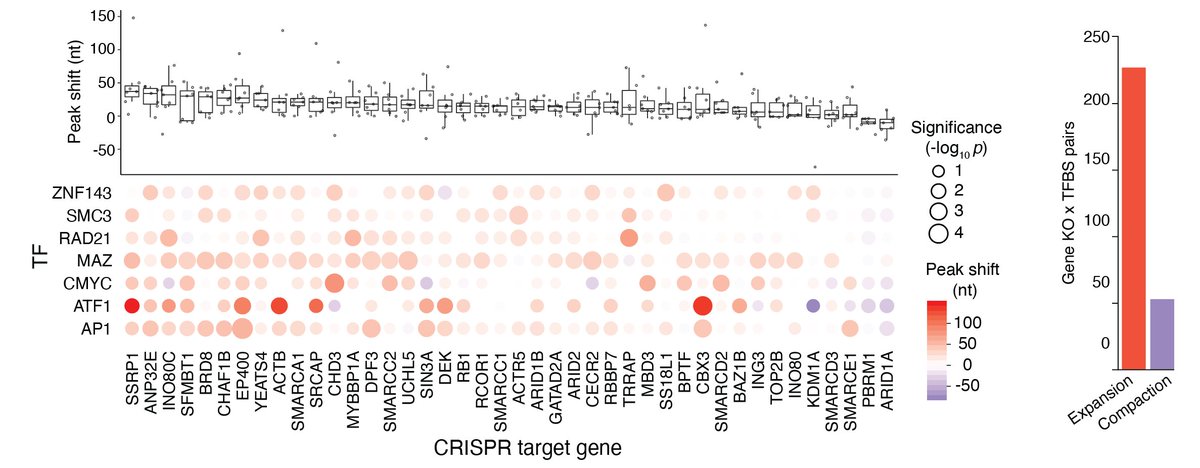
For example, ARID1A loss causes compaction at AP-1 TFBS whereas EP400 loss causes expansion at these same sites.

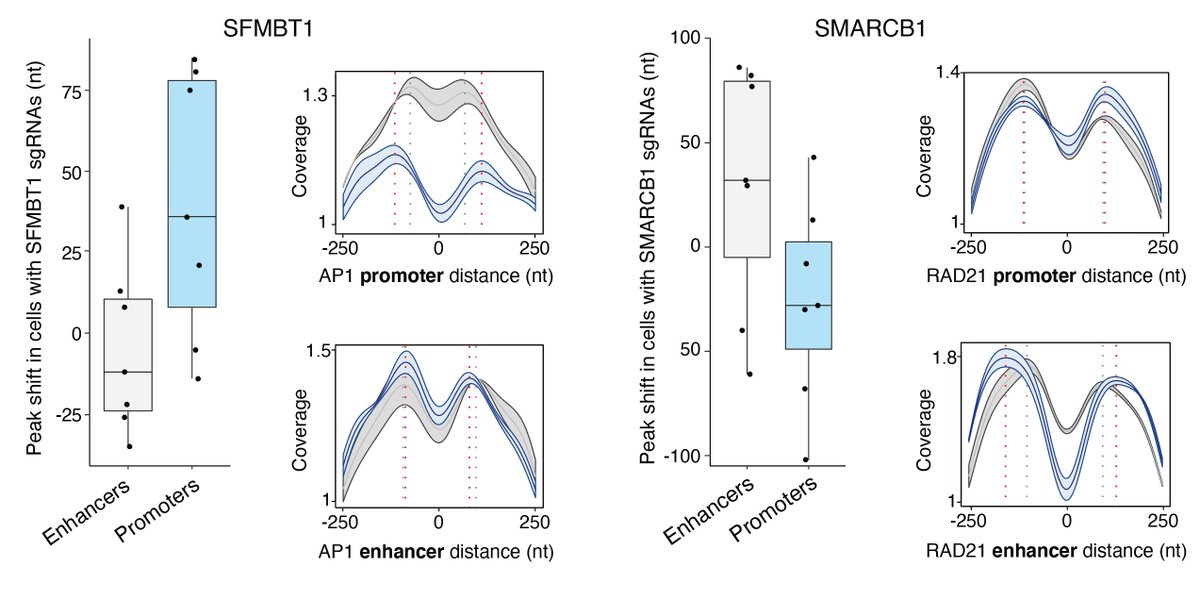
We also see effects that a specific to TFBS in promoters or enhancers. Two examples are SFMBT1 (expansion in AP-1 sites at promoters but not enhancers) and SMARCB1 (expansion in RAD21 sites at enhancers but not promoters).
I think we have toured all the highlights but there is much more in the preprint (and Supp Figs) that I encourage you to check out. This work is one of the first projects that we started in my lab in 2016-17 — with lots of great improvements suggested by colleagues & reviewers.
In addition to our A+ collaborators @NYGCTech, this work includes efforts from 5 postdocs, 2 PhD & 3 MS students. Many have moved on but made vital contributions at early stages. Incredibly proud of this team effort 🇮🇹🇮🇱🇨🇳🇲🇽🇩🇪🇺🇸🇹🇼🇹🇹🇮🇳🇦🇺 & grateful to work w/ top-notch scientists.
Several other folks @nygenome and @nyuniversity also supported us, including #MasoodZaran (now @BrookhavenLab), @satijalab, and @jamie_gregory17.
Finally, there have been several amazing recent preprints and manuscripts combining single-cell ATAC and RNA and we have been inspired by each of them.
In particular, @WJGreenleaf, @Sarah_E_Pierce, and @JeffreyGranja published a beautiful study combining CRISPR and sciATAC-seq called Spear-ATAC that elegantly uses 10x to further boost throughput. Highly recommended paper!
biorxiv.org/content/10.110…
biorxiv.org/content/10.110…
In the years to come, it will be wonderful to use technologies like CRISPR-sciATAC to understand how genetic perturbations impact genome-wide chromatin accessibility in complex systems, primary cells and patient-derived cells. A lot of exciting work ahead! 🙂
• • •
Missing some Tweet in this thread? You can try to
force a refresh