
Out today: two academic publications (not yet peer reviewed) that formally test whether the new B.1.1.7 variant is more transmissible. Both conclude yes, about 50% more. 🧵
First, a pre-print led by @erikmvolz and @neil_ferguson at Imperial, which applied a variety of different models using both genome sequence data and the S-gene dropout data I've mentioned before. imperial.ac.uk/mrc-global-inf…
Comparing genomes (sparse and lagged) and S-gene dropout (dense and up-to-date) shows the same rapid expansion we all know about in London, the East and the Southeast. 
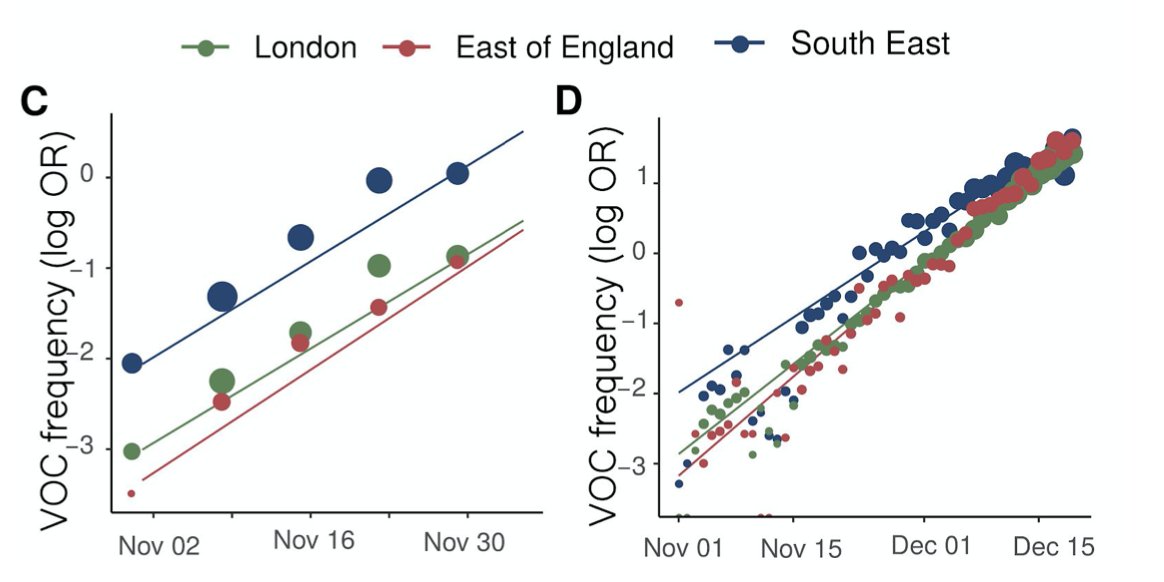
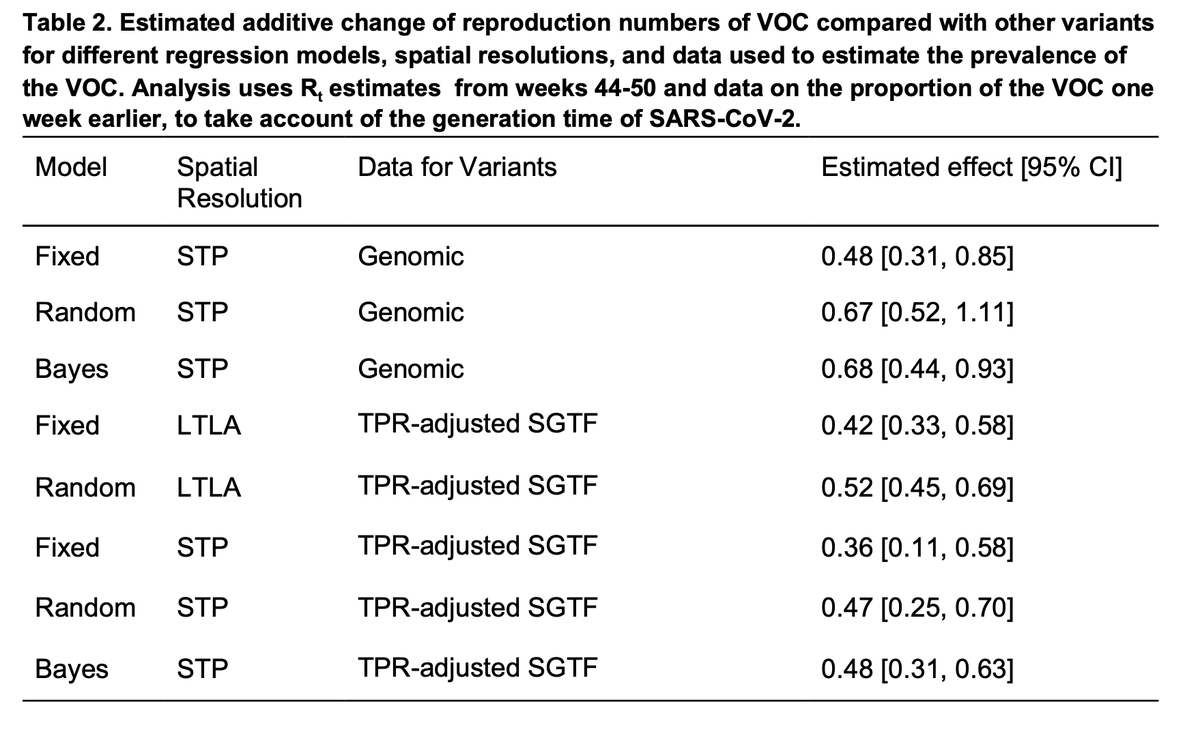
Using different models (lots more detail in the paper), spatial resolutions, and either the genomes or S-gene dropout to track B.1.1.7, the conclusion is very consistent: about a 0.5 additive increase to R.
Second, a preliminary report led by @MoritzGerstung and
@harald_voeh, who adapted a hierarchical Bayesian model they made to study the rate of positive tests around England to consider separately B.1.1.7 and other lineages. virological.org/t/lineage-spec…
@harald_voeh, who adapted a hierarchical Bayesian model they made to study the rate of positive tests around England to consider separately B.1.1.7 and other lineages. virological.org/t/lineage-spec…
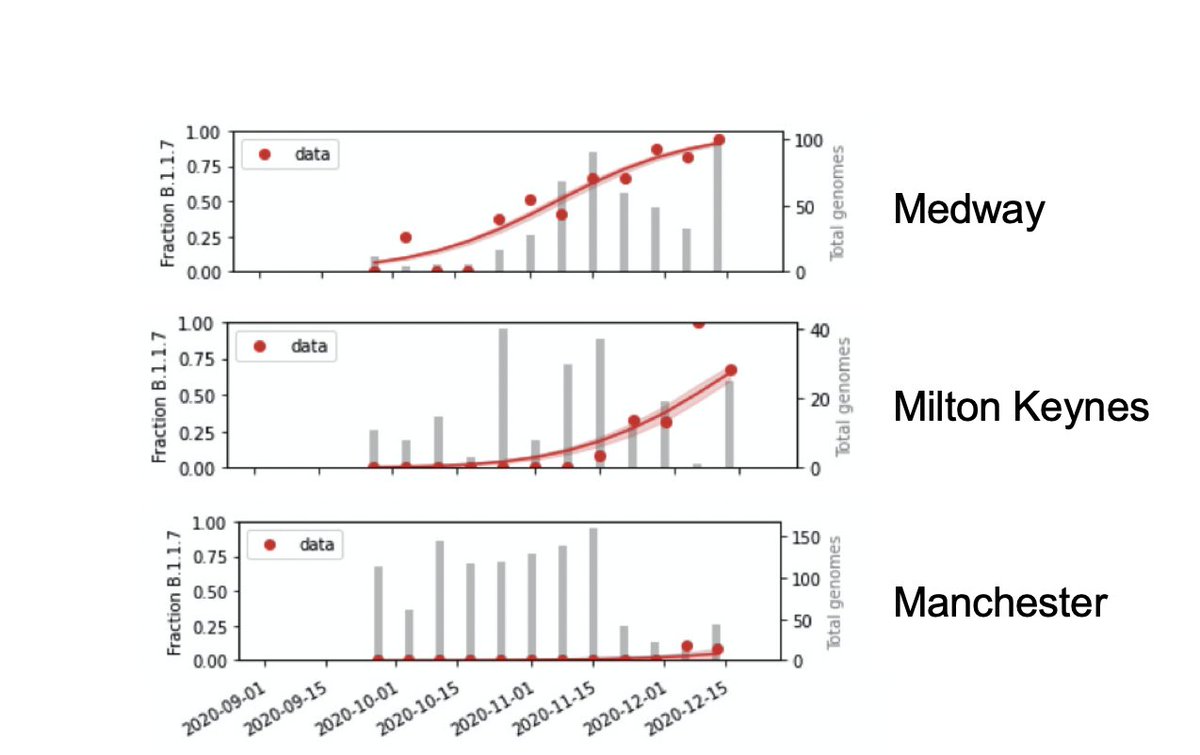
We estimate proportion of B.1.1.7 in each region of England week-by-week from ~random genome sequences from the past few month. It rose early in the SE (e.g. Medway), later elsewhere (Milton Keynes), or is just arriving (Manchester).
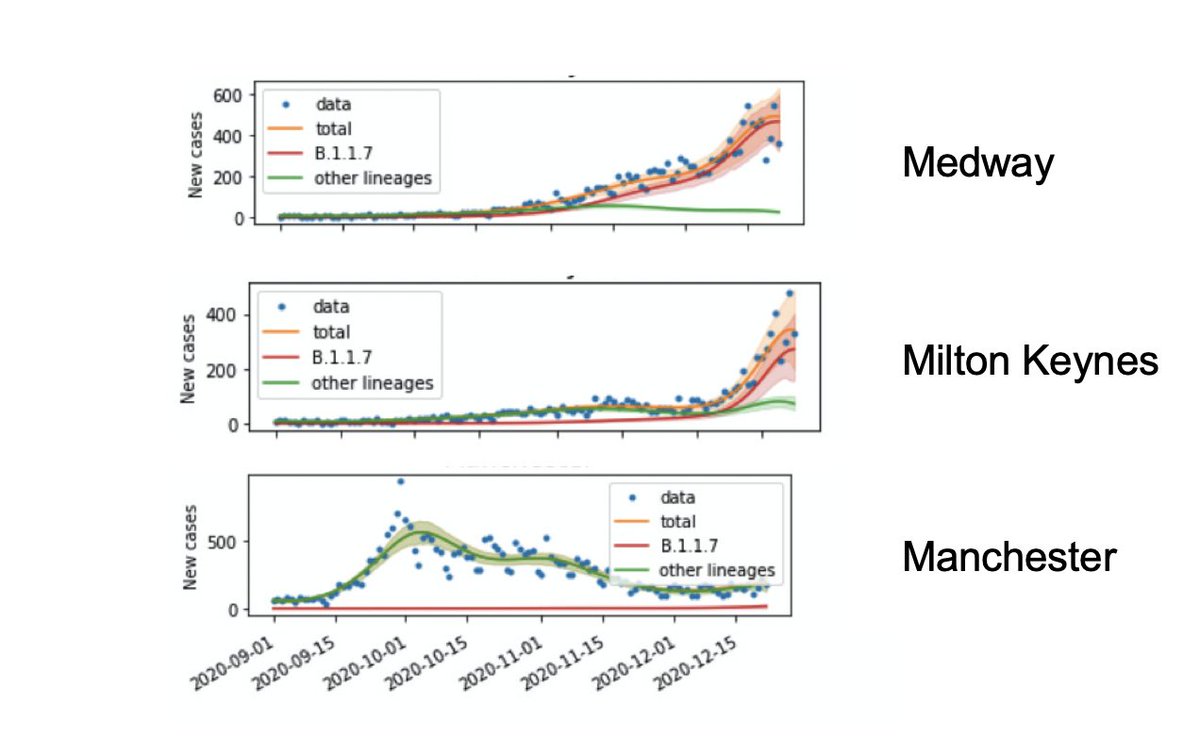
Then we use those estimates to split the observed positive test rates each day into B.1.1.7 and other lineages, so we can study their different growth patterns over time.
Finally, as in the other paper, we can model the R number. If > 1 the epidemic is growing, < 1 shrinking. And consistently (a) B.1.1.7 is 50% higher than others and (b) it was often > 1 during the November lockdown in England.
In both papers, if we look across all English regions, in the overwhelming majority B.1.1.7 expands during lockdown while other lineages contract. We almost never see the reverse. Conditions during lockdown were usually enough to suppress older variants, but not B.1.1.7.
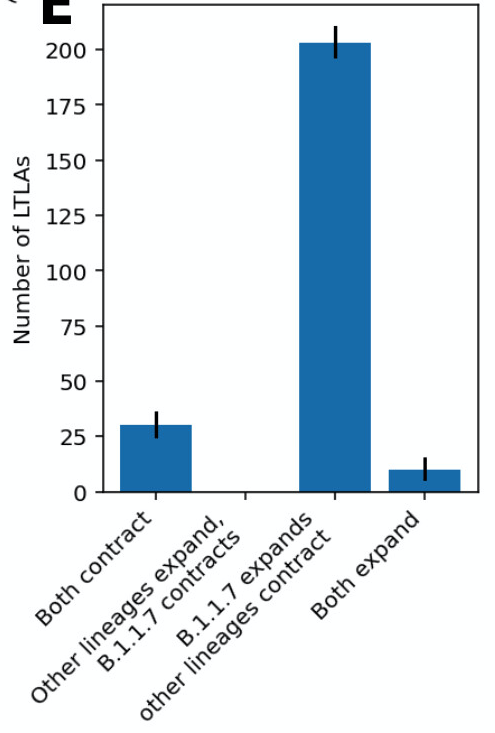
When you get consistent answers from different statistical models, that's usually a good sign that the findings are robust. As B.1.1.7 is detected around the world, I hope these analyses will be useful for countries confronting this more rapidly spreading variant.
• • •
Missing some Tweet in this thread? You can try to
force a refresh





