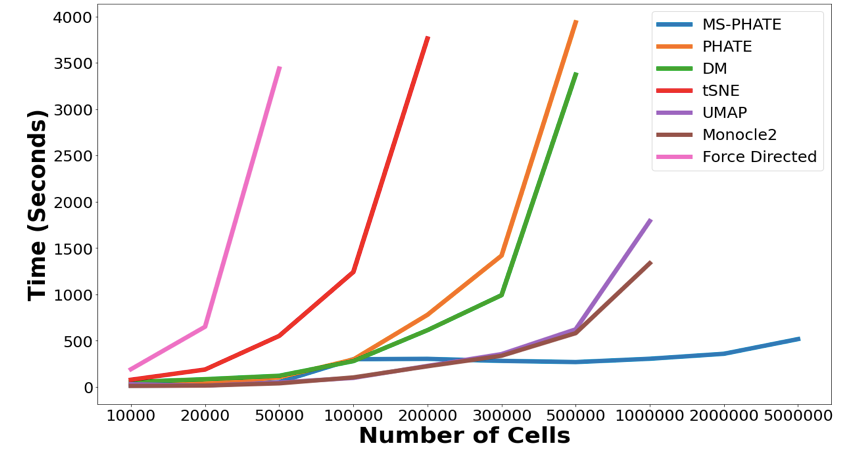
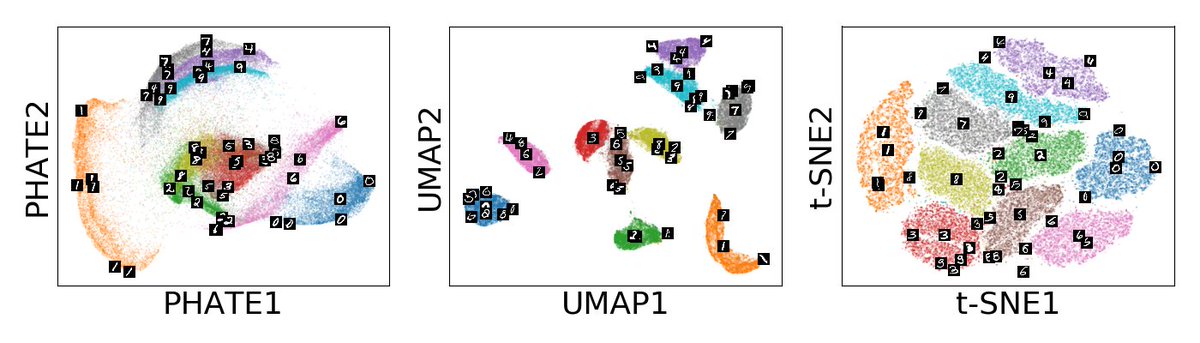
Today, I’m proud to share our latest work published in @NatureBiotech describing MELD, a #MachineLearning algorithm for #SingleCell perturbation analysis.
Read this #tweetorial to learn about the work led by @dbburkhardt and Jay Stanley 🥳🎉🧪
nature.com/articles/s4158…
(1/16)
Read this #tweetorial to learn about the work led by @dbburkhardt and Jay Stanley 🥳🎉🧪
nature.com/articles/s4158…
(1/16)
Before we get into the details of the paper, I want to give a shout out to our excellent collaborators: @david_van_dijk, Guy Wolf, @giraldezlab, and Kevan Herold. This work was possible thanks to countless discussions, experimental support, and input along the way (2/16)
I also want to share a link to software for the paper. @dbburkhardt with help from @scottgigante wrote a Python 🐍 package following the @scitkit_learn API. It’s available right now on GitHub and PyPi. Couldn't be easier:
$ pip install meld
github.com/KrishnaswamyLa…
(3/16)
$ pip install meld
github.com/KrishnaswamyLa…
(3/16)
This work was inspired by an emerging trend in single cell experiments. Moving beyond profiling tissue heterogeneity, we found labs starting to use scRNA-seq as an experimental readout.
But, how should you quantify the effect of a perturbation on a single cell dataset?
(4/16)
But, how should you quantify the effect of a perturbation on a single cell dataset?
(4/16)

We found standard tests such as differential expression between samples failed to recover differences in pathways known to play a role in the biological process under investigation. Here are 6 genes implicated in TCR stimulation for control and treatment samples
(5/16)
(5/16)
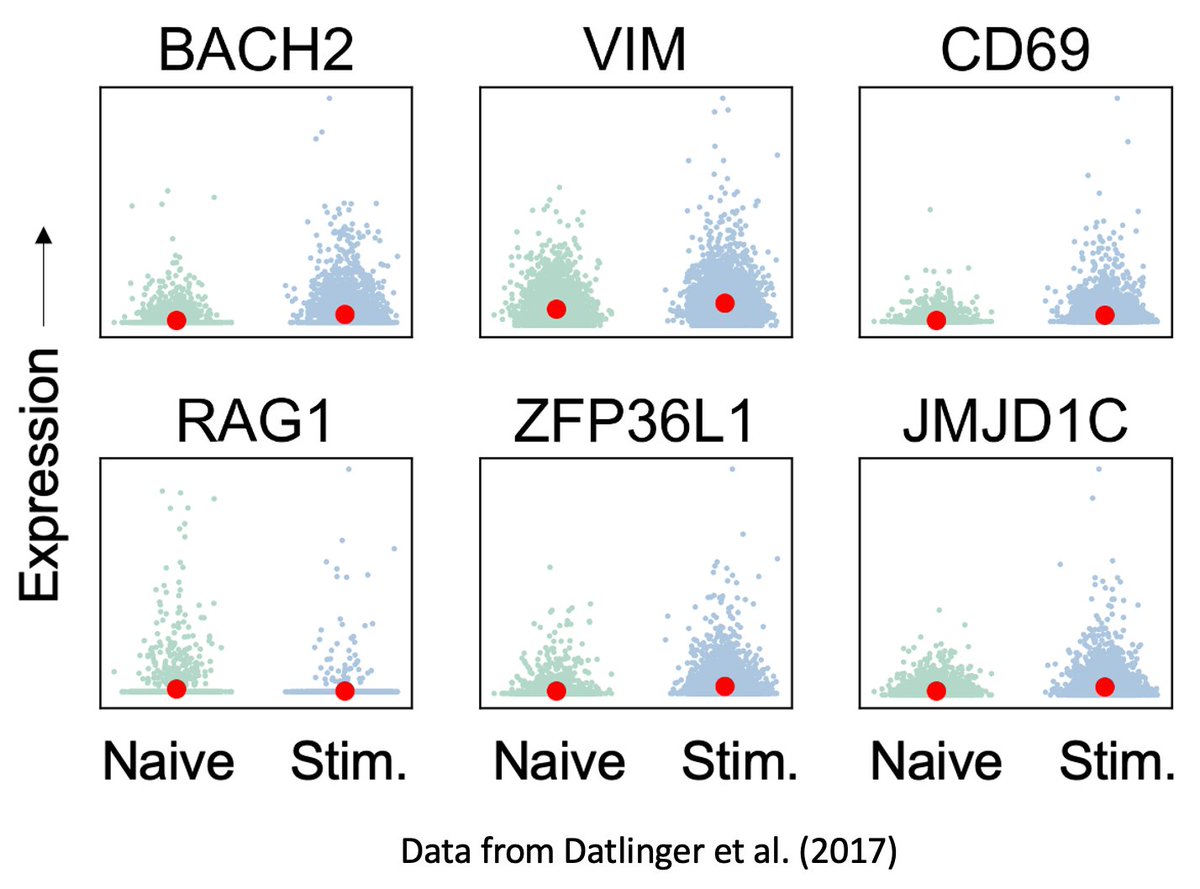
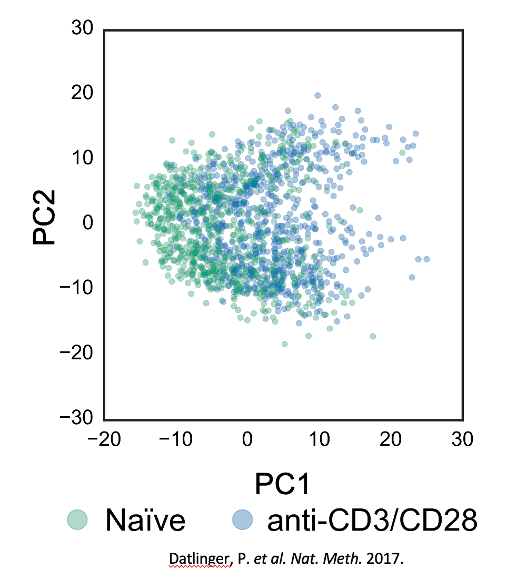
Examining these datasets, we conclude that the effect of a perturbation is often small with respect to biological heterogeneity & technical noise.
This is unsurprising. An expt treatment may only affect a few hundred genes out of the 20,000+ expressed in a given cell
(6/16)
This is unsurprising. An expt treatment may only affect a few hundred genes out of the 20,000+ expressed in a given cell
(6/16)
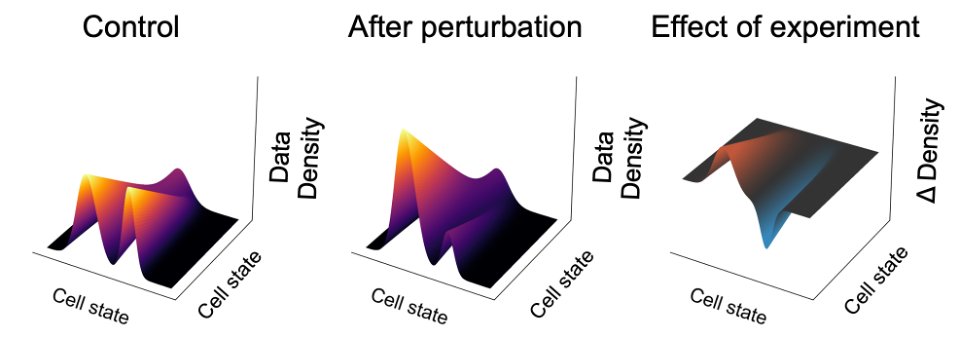
To solve this problem, we devised a new paradigm for quantifying the effect of a perturbation as a change in data density over a cell state manifold.
This approach allows us to quantify the experimental effect at the resolution of single cells, instead of clusters
(7/16)
This approach allows us to quantify the experimental effect at the resolution of single cells, instead of clusters
(7/16)
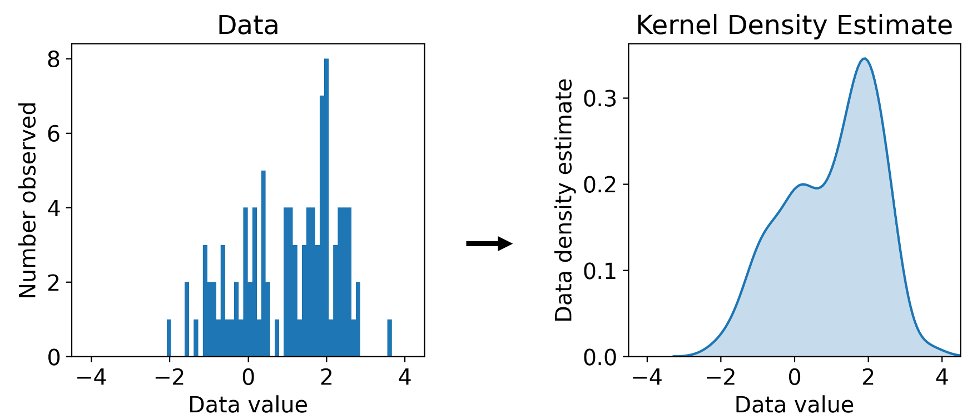
A standard approach to estimating density is using a kernel density estimator (KDE) using e.g. a Gaussian kernel. You’ve used this algorithm if you’ve ever tried making a KDE plot.
(8/16)
(8/16)
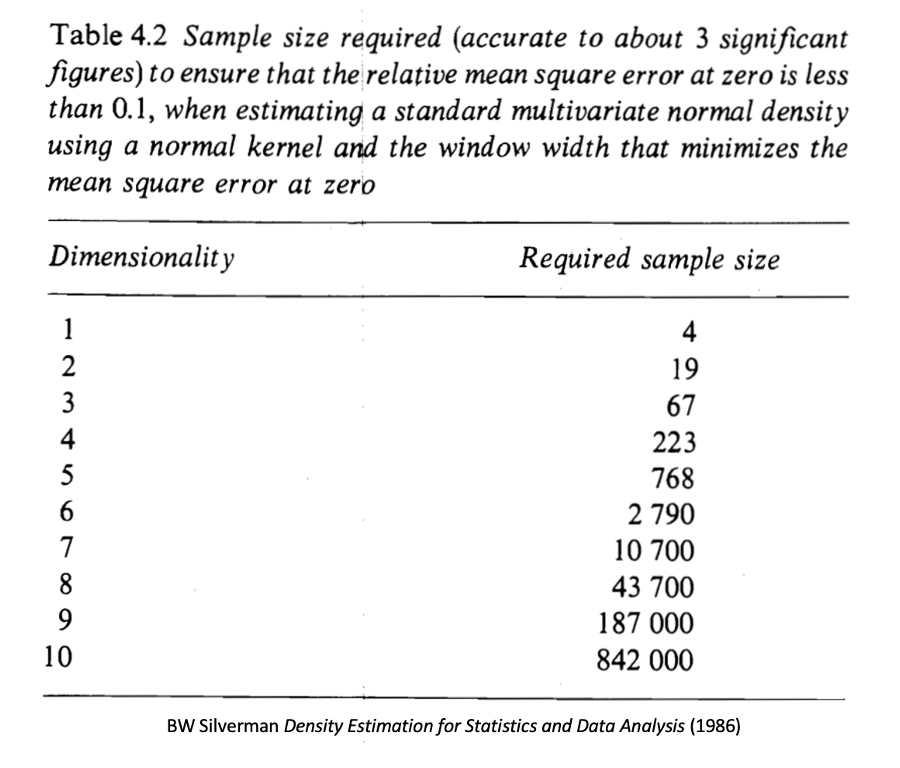
However, it's difficult to perform multivariate KDE in high dimensions. As dimensionality of the data increases, the number of observations required for accurate estimation rises exponentially.
(9/16)
(9/16)
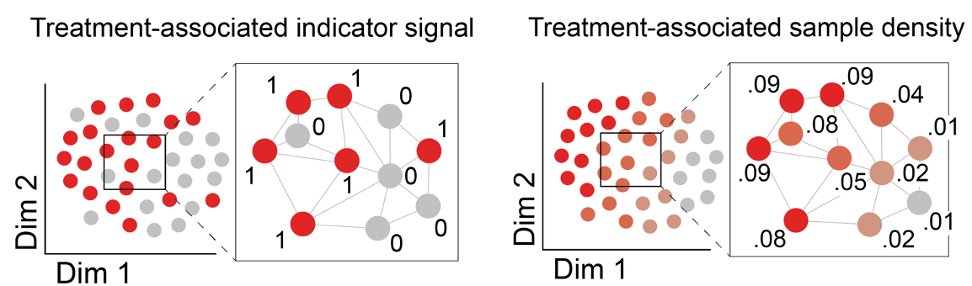
Solution: instead of estimating density in the ambient gene space, we do KDE over a cell similarity graph derived from all cells from all samples.
Using graph signal processing, we develop a heat kernel to estimate density of each sample over the graph.
(10/16)
Using graph signal processing, we develop a heat kernel to estimate density of each sample over the graph.
(10/16)
We then use the probability density of each sample to quantify the relative likelihood that each cell would be observed in the one condition relative to the others.
Here are 27K cells from the zebrafish embryo w/ or w/o Cas9 chordin KO. Higher values mean more enriched
(11/16)
Here are 27K cells from the zebrafish embryo w/ or w/o Cas9 chordin KO. Higher values mean more enriched
(11/16)
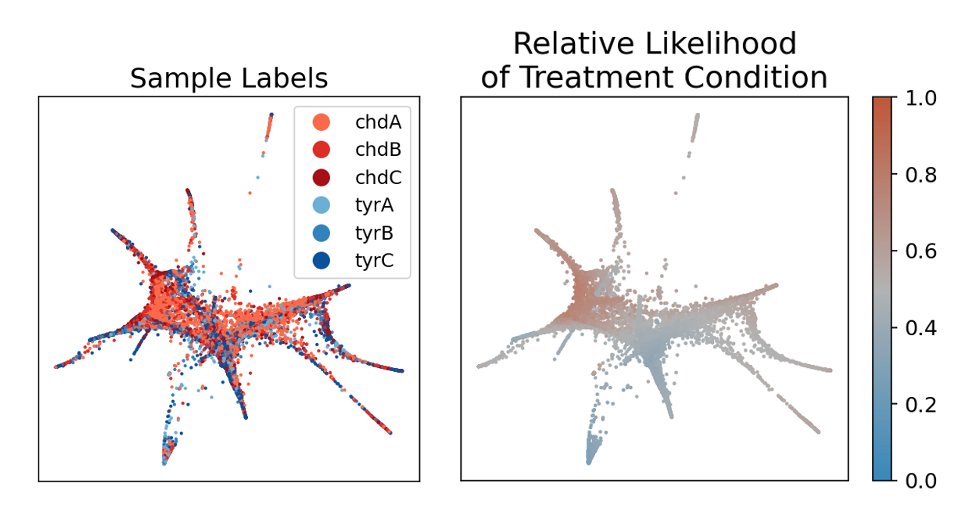
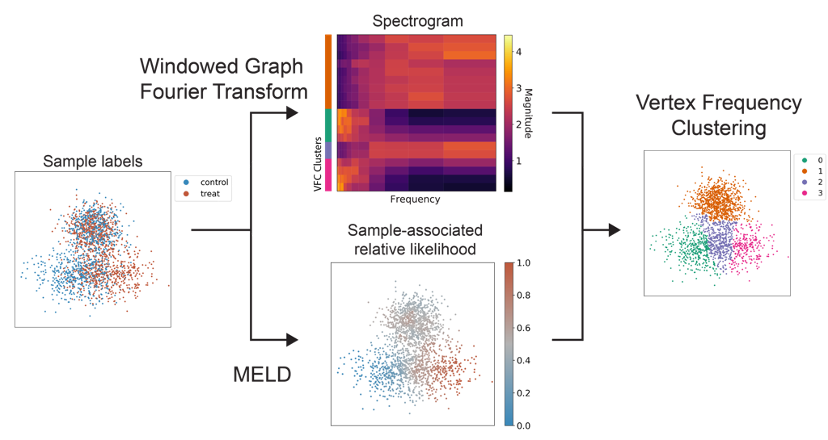
We next introduce a new clustering tool called Vertex Frequency Clustering. Using the frequency of the input sample labels and the MELD relative likelihood, we can identify cells that are transcriptionally similar AND have uniform response to a perturbation.
(12/16)
(12/16)
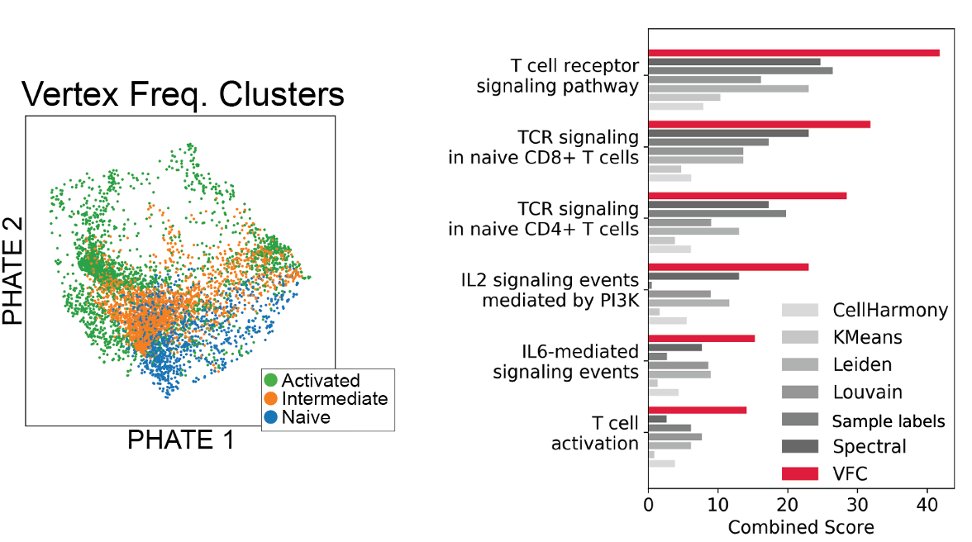
We show that using MELD + VFC, we can derive more accurate gene signatures for a perturbation compared to geometry-based clustering or simple differential expression testing!
(13/16)
(13/16)
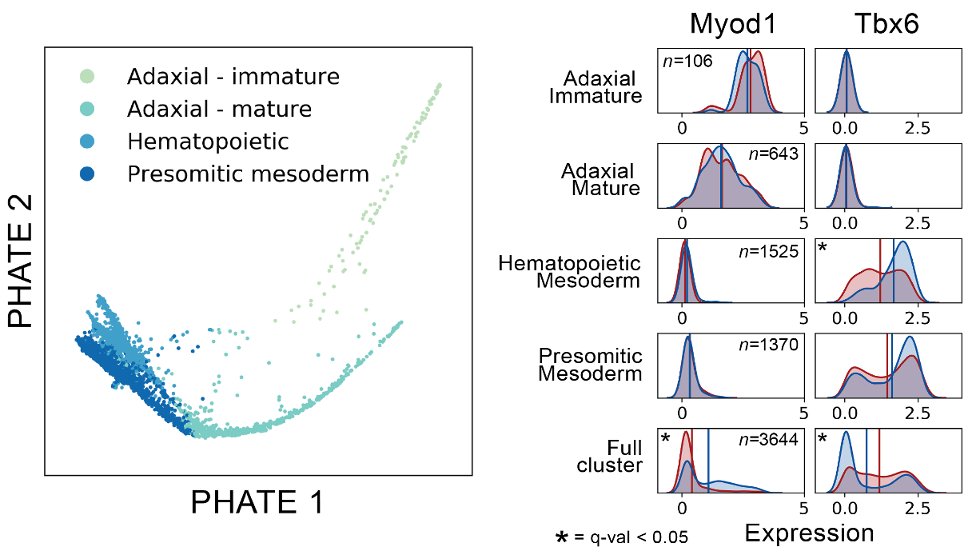
We also show that with single-cell resolution, we can reveal responses obscured by traditional cluster analysis. These cells were called a single cluster in the original analysis, but MELD uncovered divergent responses in subpopulations within the cluster 🧐
(14/16)
(14/16)
A few other methods (DA-seq, MILO, and scCODA) attempt to quantify experimental perturbations.
I’m excited to share that we are actively collaborating with those groups on an open-source benchmark. You can see our conversations here: github.com/singlecellopen…
(15/16)
I’m excited to share that we are actively collaborating with those groups on an open-source benchmark. You can see our conversations here: github.com/singlecellopen…
(15/16)
Congrats for getting this far! Thanks to @dbburkhardt for help writing this tweetorial. We’re excited to see how you use MELD in your work 🥳
(16/16)
(16/16)
• • •
Missing some Tweet in this thread? You can try to
force a refresh